
Deficit fosfomanomutázy 2: klinická, biochemická a molekulárně-genetická charakteristika 22 pacientů diagnostikovaných v České republice
Authors:
A. Čechová; N. Ondrušková; M. Tesařová; H. Hansíková; J. Zeman; T. Honzík
Authors‘ workplace:
Klinika dětského a dorostového lékařství 1. LF UK a VFN, Praha
Published in:
Čes-slov Pediat 2018; 73 (6): 365-374.
Category:
Overview
Úvod:
Deficit fosfomanomutázy 2 (PMM2-CDG) je nejčastějším typem poruch N-glykosylace s popsanými >900 pacienty. Onemocnění je autosomálně recesivně dědičné a biochemickou podstatou je porucha přeměny manóza-6-fosfátu na manóza-1-fosfát (M1P). Onemocnění se projevuje encefalopatií, neuropatií, typickou dysmorfií, atrofií mozečku a koagulopatií. Práce prezentuje výsledky klinických, biochemických a molekulárních vyšetření pacientů diagnostikovaných v ČR.
Výsledky:
Od roku 2002 bylo v ČR diagnostikováno 22 pacientů z 18 rodin. Dvě děti zemřely v kojeneckém věku. Věk žijících 20 pacientů je v rozpětí 9 měsíců až 29 let (medián 14 let). U všech pacientů je přítomný hypotonický a mozečkový syndrom, strabismus, deformity skeletu, koagulopatie a mentální retardace, která je v pásmu od lehkého až po hluboké postižení. U 94 % pacientů jsme prokázali atrofii mozečku. Typická dysmorfie (atypické rozložení tuku a vpáčení bradavek) byla zjištěna u 82 % dětí. U 9 pacientů se projevila epilepsie a 3 pacienti prodělali iktu podobné příhody. Isoelektrická fokusace transferinu v séru prokázala u všech pacientů zvýšené zastoupení nízkosialovaných forem. Diagnóza byla potvrzena enzymologicky detekcí snížené aktivity PMM2 v lymfocytech či fibroblastech a/nebo molekulárně-geneticky. V našem souboru pacientů bylo zastoupeno 10 mutací v genu PMM2, všichni pacienti jsou složení heterozygoti a 71 % mutovaných alel neslo jednu ze dvou prevalentních patogenních variant (c.422G>A, c.338C>T).
Závěr:
S ohledem na odhadovanou incidenci PMM2-CDG 1 : 20 000 se jedná o onemocnění v ČR poddiagnostikované. PMM2-CDG patří do diferenciální diagnostiky všech dětí s atrofií mozečku, a to i bez přítomnosti charakteristických dysmorfických rysů. V plánu je zařazení českých pacientů do prospektivní multicentrické mezinárodní studie hodnotící přirozený průběh onemocnění a eventuálně zařazení do klinické studie s novou experimentální léčebnou molekulou LipoM1P (manóza-1-fosfát inkorporovaná do liposomu).
KLÍČOVÁ SLOVA:
CDG syndrom, fosfomanomutáza 2, PMM2-CDG, isoelektrická fokusace, atrofie mozečku, koagulopatie
ÚVOD
Glykosylace je jednou z nejběžnějších a nejkomplexnějších posttranslačních úprav proteinů a lipidů [1], která se týká více než poloviny buněčných bílkovin [2]. Odhaduje se, že na syntéze glykokonjugátů se podílí >2 % našich genů [2]. Její procesy se odehrávají v cytoplazmě, endoplazmatickém retikulu a Golgiho aparátu. Glykosylace ovlivňuje stabilitu, konformaci, lokalizaci a odolnost proteinů vůči proteázám, podílí se na buněčných interakcích, účastní se vazby cytoskeletu s extracelulární matrix [3]. Glykoproteiny se podle typu vazby připojující oligosacharid (glykan) dělí na N-, O- a C-vázané glykoproteiny; z dalších glykosylovaných sloučenin zmiňme proteoglykany, glykosfingolipidy a proteiny s glykosylfosfatidylinositolovou (GPI) kotvou.
Dědičné poruchy glykosylace (Congenital Disorders of Glycosylation, CDG) jsou širokou skupinou geneticky podmíněných onemocnění. Od svého objevení v roce 1980 [4] se podařilo popsat více než 125 typů glykosylačních poruch [5]. Vzhledem k velké rozmanitosti glykosylovaných molekul zahrnují i CDG syndromy široké spektrum klinických projevů od monosymptomatického postižení po těžké multiorgánové dysfunkce a fetální hydrops [6].
Základem diagnostiky pro poruchy N-glykoproteinů je isoelektrická fokusace (IEF) transferinu [7] a pro defekty O-glykoproteinů IEF apolipoproteinu CIII [8], dále je využívaná analýza celkových glykanů v séru/plazmě hmotnostní spektrometrií [9]. Potvrzení diagnózy probíhá na úrovni průkazu snížené aktivity postiženého enzymu a/nebo molekulárně-geneticky průkazem patogenní mutace v příslušeném genu. Od dřívějšího dělení N-glykosylačních poruch na CDG typ I (porucha syntézy oligosacharidu vázaného přes lipid dolichol lokalizovaná v cytoplazmě a endoplazmatickém retikulu) a II (porucha modifikace N-glykoproteinů v Golgiho aparátu) [10] se dnes upouští a jednotlivá onemocnění se nazývají podle postiženého enzymu/genu [11]. Léčba je zatím známá jen u několika typů CDG, a to buď ve formě perorální suplementace (např. galaktóza u PGM1-CDG, manóza u MPI-CDG), nebo transplantací orgánů (např. játra u MPI-CDG, srdce u DOLK-CDG) [12].
Deficit fosfomanomutázy 2 (PMM2-CDG) je nejčastějším typem poruch N-glykosylace s popsanými >900 pacienty na světě. Biochemickou podstatou onemocnění je porucha přeměny manóza-6-fosfátu na manóza-1-fosfát v cytoplazmě. Manóza-1-fosfát je prekurzor GDP-manózy a dolichol-fosfát-manózy, které slouží jako donor manózy v endoplazmatickém retikulu pro syntézu oligosacharidu vázaného přes lipid (LLO). Onemocnění se projevuje obvykle v kojeneckém věku encefalopatií, neuropatií, typickou dysmorfií, atrofií mozečku a koagulopatií. Prognóza tohoto onemocnění může být závažná (až 15–20 % dětí umírá do 2 let věku), účinná léčba dosud není známá, experimentální terapie manóza-1-fosfátem inkorporovaným do liposomu (LipoM1P) je v předfázi I/II klinické studie.
V této práci prezentujeme výsledky klinických, biochemických a molekulárních vyšetření pacientů s PMM2-CDG diagnostikovaných na Klinice dětského a dorostového lékařství 1. LF UK a VFN.
METODIKA
Pacienti
Na našem pracovišti bylo od roku 2002 s PMM2-CDG diagnostikováno 22 pacientů z 18 rodin, 13 z nich jsou dívky, 9 chlapci. Věk žijících 20 pacientů je v rozpětí 9 měsíců až 29 let (medián 14 let), dvě děti zemřely v kojeneckém věku. Všichni pacienti jsou kavkazského etnika, příbuzenské sňatky se v žádné rodině nevyskytly.
Mentální retardace (intelektuální disabilita) byla hodnocena na základě DSM-4 (Diagnostic and Statistical Manual of Mental Disorders) klasifikace Americké psychiatrické společnosti.
Trombotické/tromboembolické a krvácivé komplikace vyžadující antikoagulační léčbu definují těžkou koagulopatii.
Metody
K potvrzení defektu na úrovni biosyntézy N-glykoproteinů jsme použili isoelektrickou fokusaci sérového transferinu s následnou imunofixací dle dříve publikované metodiky [8]. Aktivita PMM2 byla měřena spektrofotometricky při 340 nm jako přeměna NADP+ na NADPH v izolovaných lymfocytech z periferní krve a/nebo v kultivovaných fibroblastech také podle dříve publikované metodiky (Hansíková H, Ondrušková N, Honzík T, Veselá K, Horová E, Švecová Š, Tesařová M, Zeman J. Aktivita fosfomanomutázy 2 u pacientů s podezřením na dědičnou poruchu glykosylace. Klin Biochem Metab 2016; 24 (sv. 45, 2): 67‒74).
Molekulárně-geneticky jsme kauzální mutace prokazovali přímou sekvencí osmi exonů a přilehlých intronových oblastí PMM2 genu. Mutace byly u všech pacientů vždy potvrzeny analýzou DNA obou rodičů.
Etika
Všechny odběry a vyšetření byly provedeny po vyjádření informovaného souhlasu zákonnými zástupci pacienta. Studie byla provedena v souladu s Helsinskou deklarací Světové lékařské asociace.
VÝSLEDKY
První projevy onemocnění
U všech našich pacientů se první projevy onemocnění objevily při narození (n = 4) či v časném kojeneckém věku (n = 18), průměrný věk při nástupu onemocnění byl 2,3 měsíce (rozptyl 0 dní – 6 měsíců).
Dva novorozenci měli při nástupu onemocnění dominantní příznaky panhypopituitarismu s iontovým rozvratem a opakovanými hypoglykémiemi. Třetí chlapec manifestoval 14. den života jaterní selhání se smíšenou koagulační poruchou a následným multi-orgánovým selháním. Odmítání kojení, opakované zvracení a alterace stavu byly u čtvrtého novorozence projevem rozvoje post-hemorhagického hydrocefalu.
Typickými projevy při manifestaci v kojeneckém věku bylo opoždění psychomotorického vývoje (n = 17), hypotonie (n = 16), strabismus (n = 11), typická dysmorfie (atypická distribuce tuku, vpáčení prsních bradavek, n = 7), neprospívání (n = 7) a mikrocefalie (n = 5). U jedné dívky byl psychomotorický vývoj při prvním vyšetření hodnocen v mezích širší normy a vyšetřovaná byla pro epizody stáčení bulbů s hypotonií a náhodným nálezem hepatopatie a hepatomegalie. Průměrná prodleva diagnostikování nemoci u našich pacientů je 4,5 roku (rozptyl 2 měsíce – 15 let), ale významně se liší podle data narození: u pacientů narozených před rokem 2000 byla prodleva 10,6 roku, u pacientů narozených po roce 2000 jen 1 rok.
Klinické projevy
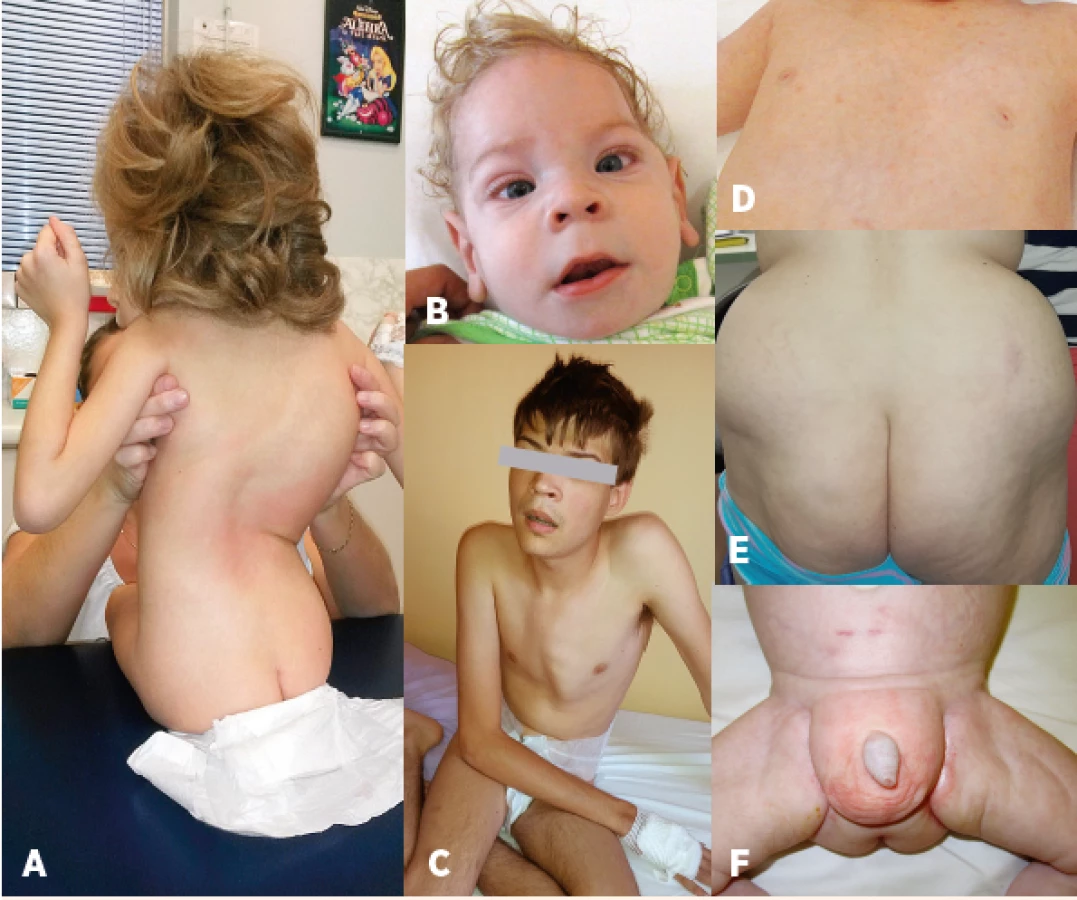
Klinické projevy a četnost jednotlivých příznaků u našich pacientů jsou shrnuty v obrázku 1. Typický fenotyp našich pacientů zahrnuje psychomotorickou retardaci, hypotonii, strabismus a typickou dysmorfii (obr. 2). Mentální retardace byla hodnocena u devatenácti dosud žijících pacientů starších 2 let: tři pacienti měli hlubokou, pět těžkou, šest středně závažnou a pět mírnou mentální retardaci. Inteligenční kvocient byl stanoven u dvanácti pacientů s maximální hodnotou 66 bodů. Mikrocefalii jsme zaznamenali v 73 % případů. Z neurologických projevů se u našich pacientů dále vyskytuje periferní neuropatie a mozečkový syndrom. Epilepsie se vyskytla u devíti pacientů, jedná se o atonické i tonicko-klonické záchvaty, první záchvat se projevil od 6 měsíců do 8 let, u jednoho pacienta jsou záchvaty vázané na horečnaté stavy. Záchvaty jsou u pěti pacientů dobře kompenzované na monoterapii, u tří pacientů kompenzované na dvojkombinaci antiepileptik, u jednoho pacienta se ani dvojkombinací léků nepodařilo docílit ideální kompenzace a 1x měsíčně se stále objevují epiparoxysmy. Iktu podobné příhody se vyskytly u tří pacientů ve věku 2–29 let (průměrný věk 11,8 roku). U dvou dívek se vyskytly opakovaně jako hemiparéza trvající několik dní až měsíc, z toho jedna z dívek se při atace chabosti 1x topila, u jednoho chlapce se vyskytla jen jedna epizoda s projevy 4 dny trvající alterace vědomí se spasticitou končetin při horečce.

Fig. 2. Characteristic phenotype: A – bone deformities – scoliosis, B – craniofacial
dysmorphism, strabismus, C – pectus carinatum, D – nipples inversion,
E – atypical supragluteal fat distribution, F – atypical perigenital fat
distribution.
Z extraneuronálních projevů jsou nejběžnější endokrinologické, gastroenterologické, oční, nefrologické a kardiologické abnormity. Z endokrinologických nálezů jsme u našich pacientů nejčastěji pozorovali hypothyreózu (u 50 %), která je velmi často subklinická. Hypogonadismus jsme zachytili u všech sledovaných dívek (6/6) v pubertálním věku, ale nevyskytl se u žádného z chlapců (0/3) v daném věku. Sekundární pohlavní znaky se u těchto chlapců vyvinuly normálně. Z dalších endokrinologických patologií se u dvou dětí od novorozeneckého věku rozvinul panhypopituitarismus.
Dalšími častými příznaky byla růstová retardace a neprospívání (u 59, resp. 41 % pacientů). Tyto příznaky se vyvíjejí postnatálně, naprostá většina dětí má při narození normální antropometrická data. Často se vyskytují v kombinaci: 2/3 neprospívajících pacientů mají zároveň i růstovou retardaci.
Kardiologické obtíže jsme pozorovali u 38 % pacientů a zahrnovaly především vrozené vývojové vady srdce (19 %, z toho polovina významných), kardiomyopatii (10 %, z toho polovina vymizela) a nekonstantní perikardiální výpotek (10 %).
Oční patologie se vyskytla až u 85 % pacientů: kromě výše zmíněného strabismu jsme poměrně často dokumentovali retinitis pigmentosa (37 %), refrakční vady různé tíže (30 %), těžké kombinované vady (15 %, atrofie terčů zrakových nervů, katarakta) a u jednoho pacienta byla přítomná bilaterální amauróza.
Abnormální nefrologický nález byl přítomný u 45 % pacientů: nejčastěji se vyskytovala nespecifická nefromegalie se setřelou kresbou bez poruchy funkce, u tří pacientů byla zachycena signifikantní proteinurie (u jednoho pacienta se rozvinul nefrotický syndrom), u tří pacientů se vyskytla cystická přestavba ledvin a u dvou pacientů bylo postižení ledvin spojeno s obtížně kontrolovanou hypertenzí.
Výsledky zobrazovacího vyšetření mozku byly dostupné u 18 pacientů (17x MR, 1x CT), z toho u 17 dětí byl nález atrofie mozečku. U jedné dívky byl na MR mozku ve věku 1 měsíce normální nález bez mozečkové atrofie. U jednoho chlapce byla provedena magnetická rezonance dvakrát, a to ve věku 10 měsíců a téměř 6,5 let: při srovnání snímků je patrná progrese atrofizace mozečku (obr. 3).
Fig. 3. Brain MRI (T2W) showing progressive cerebellar atrophy
in a boy with PMM2-CDG: the same patient at the
age of A – 10 months, B – 6.5 years.

Klinické známky neuropatie (areflexie/hyporeflexie) byly přítomné u 12 pacientů. EMG vyšetření bylo provedeno u deseti těchto pacientů a u šesti s potvrzením axonálně-demyelinizační periferní neuropatie. Medián věku pacientů s EMG patologií byl 20 let, zatímco při fyziologickém nálezu 7 let.
Symptomy koagulopatie se vyskytly u 36 % pacientů: velmi často se jednalo o benigní nález snadnější tvorby modřin, jen u čtyř pacientů byly příznaky závažnější a byla u nich nutná dlouhodobá antikoagulační terapie. Dvě pacientky prodělaly ischemickou cévní mozkovou příhodu – u první se projevila ve věku 11 let těžkou levostrannou hemiparézou a deviací bulbů, druhá dívka měla ve věku 2,5 let pravostrannou parézu horní končetiny. U první bylo zvoleno 4měsíční antikoagulační zajištění, u druhé bylo vzhledem k současné heterozygotní mutaci pro faktor V Leiden zvoleno dlouhodobé antitrombotické zajištění. Opakované periferní žilní trombózy (více než 10 trombóz na horních i dolních končetinách) byly u třetí pacientky, která je zároveň heterozygotkou mutace pro faktor V Leiden, důvodem pro zavedení kombinované antitrombotické a antikoagulační terapie. Poslední pacient neprodělal jasnou trombotickou příhodu, ale přesto byl vzhledem k závažnému celkovému stavu a koagulační patologii přechodně zajištěn antikoagulancii.
Prognóza – mortalita
V současné době je osm našich pacientů dospělých, tři jsou v adolescentním věku, šest je školního věku a tři jsou v batolecím věku. Klinický obraz našich pacientů v dospělém věku je charakteristický především stabilizací stavu včetně úrovně intelektuální disability, neurologického nálezu a laboratorních nálezů (hladina jaterních testů a koagulačních parametrů).
Dva chlapci zemřeli v kojeneckém věku (9 a 11 měsíců), jednalo se o děti manifestující se v novorozeneckém věku, příčinou smrti byla dekompenzace jaterního postižení při probíhajícím akutním respiračním infektu a náhlé úmrtí při horečnatém infektu v domácím prostředí u chlapce s prakticky zastaveným psychomotorickým vývojem, těžkou smíšenou koagulační poruchou, hepatopatií a mikrocystózou ledvin s významnou renoparenchymatózní hypertenzí.
Laboratorní výsledky
Nejčastější laboratorní odchylky jsou shrnuty v tabulce 1; jedná se o příznaky koagulopatie, hepatopatie, myopatie, thyreopatie a hypergonadotropního hypogonadismu u adolescentních dívek.

2ALT: rozptyl 0,4–10,3 μkat/l, průměr 1,2 μkat/l; AST: rozptyl
0,4–3,9 μkat/l, průměr 1,1 μkat/l
Koagulopatie se vyskytla u všech našich pacientů: u osmnácti pacientů byla mírné a u čtyř závažnější tíže s klinickým dopadem (viz výše). Ve všech případech se jednalo o smíšenou koagulopatii, nejčastěji se alterace týkala koagulačního faktoru XI, proteinu C a S a anti-trombinu. U devíti pacientů se vyskytlo prodloužení aPTT, z toho u čtyř také prodloužené INR.
Hepatopatie se vyskytla u šestnácti pacientů, u šesti pacientů byla zachycena závažná elevace sérových trans-amináz nad desetinásobek normy. U čtyř pacientů byla náhodně zjištěná hepatopatie jedním z prvních příznaků, pro které bylo dítě vyšetřované.
Myopatie byla laboratorně zachycena u čtrnácti pacientů, nicméně elevace kreatinkinázy a myoglobinu byla u všech mírná a žádný z pacientů nikdy neprodělal ataku rhabdomyolýzy.
Thyreopatii jsme zaznamenali u jedenácti pacientů, u většiny (n = 8) se jednalo o subklinickou formu periferní hypothyreózy bez alterace fT4. Častým nálezem byla také nižší hladina TBG (u 7 z 10 vyšetřených pacientů). U dvou pacientů se jednalo o centrální hypothyreózu v rámci panhypopituitarismu. Všichni pacienti jsou na terapii levothyroxinem a v jednom případě jodidem.
Hypergonadotropní hypogonadismus byl prokázán u všech šesti dívek v pubertálním věku s klinickými známkami pubertas tarda. Tři postpubertální chlapci měli normální hladiny pohlavních hormonů a známky rozvoje puberty.
Denzitometrie prokázala u všech pacientů (n = 9), u kterých byly výsledky dostupné, sníženou kostní denzitu. U čtyř pacientů byly dostupné výsledky celotělové denzitometrie s průměrným Z-skóre -2,8 (rozptyl -2 až -3), u ostatních pacientů byl výsledek celotělové denzitometrie hodnocen jako nevalidní. U osmi pacientů byla dostupná denzitometrie bederní oblasti s průměrným Z-skóre -3,4 (rozptyl -1,1 až -6,8), nejvýraznější byl nález u 10leté dívky s těžkou formou nemoci s výraznými kostními deformitami a trvale upoutanou na lůžko.
Biochemické a molekulárně-genetické vyšetření
U všech pacientů v souboru byl pomocí IEF nalezen patologický profil sialovaných forem sérového transferinu (TRF), konkrétně snížený tetrasialo-TRF a zvýšený obsah disialo - a asialo-TRF, typický profil CDG typu I. Ukázka profilu typu I je uvedena na obrázku 4.
K potvrzení diagnózy při pozitivní IEF TRF byla provedena enzymologická a/nebo molekulárně-genetická analýza. Snížená aktivita PMM2 pod 30 % průměru kontrol byla prokázána v izolovaných lymfocytech u 15 ze 17 a v kultivovaných fibroblastech u 6 z 8 testovaných vzorků (tab. 2). Molekulárně-genetické vyšetření u všech dětí potvrdilo diagnózu průkazem mutace v PMM2 genu. 71 % mutovaných alel nese jednu ze dvou nejčastějších patogenních variant (tab. 2). Nebyla nalezena žádná korelace mezi klinickým fenotypem, reziduální enzymatickou aktivitou a genotypem.

DISKUSE
Klinický průběh
Celkový průběh onemocnění u 22 pacientů diagnostikovaných na naší klinice se významně neliší od průběhu popsaného v literatuře [13]. Většina našich pacientů postupně rozvíjí příznaky typické pro čtyři jednotlivá stadia onemocnění [13]: multisystémové onemocnění v kojeneckém a batolecím věku, stadium ataxie a mentální retardace v dětství, stadium neuropatie a svalové atrofie v adolescentním věku a hypogonadální období v dospělosti [14]. Skupina pacientů s neonatálním nástupem onemocnění a závažnými multisystémovými příznaky je spojena s vysokou mortalitou až 20 % [15], v naší skupině pacientů došlo k úmrtí 2/4 těchto pacientů.
Prenatální projevy a typická dysmorfie
Prenatální projevy (hydrops plodu, hydrops placenty, polyhydramnion), které jsou popisované v literatuře jen vzácně [16], se u našich pacientů nevyskytly. Kraniofaciální dysmorfii jsme u našich pacientů pozorovali často (71 %), ale byla povětšinou velmi mírná. Z vrozených malformací se vyskytuje kryptorchismus (u 50 % našich pacientů) a vrozené srdeční vady (19 % našich pacientů).
Atypická distribuce tuku a s tím spojené vpáčené bradavky (podmíněné depozitem tuku při minimu glandulární tkáně v oblasti mamily) je přítomna u 25–100 % pacientů s PMM2-CDG dle jednotlivých studií, u našich pacientů nebyla přítomná pouze u čtyř dětí. Etiopatogeneze atypického ukládání tuku není zcela jasná, nicméně předpokládá se souvislost se zkráceným biologickým poločasem IGF-1 a hypoglykosylací inzulinových a leptinových receptorů s následným ovlivněním adipogeneze [17]. Vpáčené bradavky jsou popisovány i jednostranně [16], toto jsme zaznamenali u 2 pacientů. S věkem má tento nález tendenci regredovat [18]. V našem souboru vpáčené bradavky vymizely u 5 pacientů (tj. 26 %), atypická depozita tuku u 6 pacientů (tj. 40 %).
Neurologické příznaky
PMM2-CDG se může projevovat izolovanými neurologickými projevy či jako multiviscerální forma. Neurologická manifestace nejčastěji zahrnuje psychomotorickou retardaci, mentální retardaci (intelektuální disabilitu), hypotonii, mozečkový syndrom a periferní neuropatii. Psychomotorická retardace se vyskytuje u naprosté většiny pacientů, včetně pacientů naší kohorty. V literatuře bylo dosud popsáno jen 20 pacientů bez psychomotorické retardace a s normálním IQ [15, 19–24]. Intelektuální disabilita byla v naší skupině posunuta směrem k těžším formám – zastoupení hraničního intelektu (podprůměr) či normální inteligence nebylo v naší skupině pozorováno, naopak hluboká mentální retardace byla u 23 % našich pacientů. Periferní neuropatie se vyskytla u 63 % pacientů, i když jen u 2/3 vyšetřených pacientů byl nález potvrzen na EMG. Rozvíjí se obvykle v druhé dekádě života: popisujeme ho u 85 % pacientů starších 10 let, ale jen u 17 % mladších 10 let. Málo časté jsou poruchy hybnosti: u několika málo pacientů byly popsané dystonické dyskinetické pohyby rukou či cervikální dystonie [25, 26]. Žádné podobné obtíže se u našich pacientů nevyskytly.
Z akutních neurologických projevů se nejčastěji vyskytují epilepsie a iktu podobné příhody, oboje s poměrně nízkou incidencí (dokumentována pouze u 68 resp. 36 pacientů v literatuře). Křeče se mohou objevit i v neonatálním období [27], což se mezi našimi pacienty neobjevilo. Mohou mít charakter tonicko-klonický i atonický a u naprosté většiny pacientů jsou dobře kompenzované na antiepileptické terapii. Za nejčastější spouštěč ataky iktu podobných příhod se považuje febrilní infekt a trauma hlavy [28]. Předpokládá se, že patofyziologickým podkladem by mohla být kanálopatie podobně jako u CACNA1A – asociované familiární hemiplegické migrény s hypoglykosylací týchž napěťově řízených CaV2.1 kanálů [29]. V naší skupině se iktu podobné příhody vyskytly u tří pacientů: u dvou pacientů k atakám došlo v dětském věku, ale jedna pacientka prodělala opakované ataky v adolescenci a dokonce v dospělosti, což je v kontrastu s literaturou [30].
Atrofie mozečku je také velmi častým nálezem (94 % našich pacientů) a je možno ji detekovat i prenatálně [31] či v novorozeneckém věku [24]. Přesto se nejedná o hypoplazii, neboť zmenšení objemu mozečku má progresivní charakter v časném dětském věku [32]. To se v naší skupině podařilo prokázat zobrazením mozku téhož pacienta ve věku 10 měsíců a 6,5 roku (obr. 3). U jednoho pacienta byl na MR mozku ve věku 1 měsíce normální nález na mozečku, což můžeme vysvětlit časným věkem, ve kterém bylo vyšetření provedeno (srovnávací snímek v pozdějším věku bohužel není dostupný), jak bylo popsáno v některých pracích [20, 21].
Strabismus a pigmentová degenerace sítnice, dokumentovaná u 85 % resp. 22 % z 535 PMM2 pacientů z literatury s popisem očních projevů, byly nalezeny v našem souboru u 81 % resp. 41 % jedinců. Neuropatie optiku je vzácnějším projevem PMM2-CDG s popsaným pouze jedním pacientem s poruchou zrakové ostrosti v tíži praktické slepoty.
Viscerální postižení
Ledvinné postižení je součástí většinou těžkých multiviscerálních fenotypů PMM2-CDG a bylo popsáno u 85 % z 56 pacientů z literatury s důkladnějším popisem renálních funkcí. Charakteristickým nálezem je cystická dysplazie ledvin, tubulární proteinurie, méně často hydronefróza. Nefrotická proteinurie, přítomná u jednoho našeho chlapce, byla dokumentována ve světě pouze u šesti pacientů [24, 33–36].
Jaterní selhání bylo příčinou úmrtí u našeho 11měsíčního chlapce s PMM2-CDG. Hepatopatie se dále vyskytla u šestnácti pacientů, u šesti z nich byla zachycena závažná elevace sérových transamináz s přechodnou poruchou jaterních funkcí. Těžké jaterní onemocnění v kojeneckém věku je většinou asociováno s multiviscerálním fenotypem. Autopsie v publikovaných případech ukázala cholestázu s dilatací dobrých žlučovodů, periportální fibrózu, steatózu až po portoportální přemosťující fibrózu anebo cirhózu [13, 31, 37–42]. I u mírnějších fenotypů onemocnění je hepatomegalie a hepatopatie přítomná u velkého počtu pacientů (12,5–100 %) [18, 30, 43, 44], nejedná se však o progresivní jaterní onemocnění. Jaterní testy výrazněji stoupají v době horečnatých stavů, nicméně laboratorní i klinická patologie má tendenci se mírnit či dokonce ustupovat po druhé dekádě života. Jaterní biopsie je zřídkakdy u těchto pacientů prováděna a ukazuje steatózu [23, 45, 46] či lysosomální inkluze v hepatocytech [47].
Endokrinopatie
Mezi charakteristické endokrinologické abnormity u pacientů s PMM2-CDG patří postnatální růstová porucha, thyreopatie, opožděná puberta, hypoglykémie a osteo-poróza. Více než 50 % našich pacientů má významnou růstovou poruchu, což je v souladu s literaturou, kde 49 % pacientů z 53 hodnocených subjektů v různých studiích se prezentovalo postnatálním růstovým selháním. Pacienti mají normální či dokonce vyšší koncentraci růstového hormonu v krvi, ale snížená je koncentrace IGF-1, IGFBP3 a acid-labile subunit (ALS) [45, 48]. IGF-1 není sice glykosylován, ale více než 90 % IGF-1 molekul je vázaných na transportní glykoproteiny IGFBP3 a ALS. Jejich abnormní glykosylace vede ke zkrácenému biologickému poločasu IGF-1. Jen jedna práce dokládá úspěšnou terapii rekombinantním lidským IGF-1 [49]. Abnormální thyreoidální funkce byla nalezena u 255 jedinců s PMM2-CDG v literatuře. Podobně jako u našich pacientů, většinou se jednalo o subklinickou formu periferní hypothyreózy na základě měření hladiny TSH. Částečná deficience hypoglykosylovaného TBG je přítomná u 75 % pacientů při zkrácení jeho biologického poločasu na 15 %, nicméně jeho deficit nejspíše nevede k poruše thyroidálních funkcí [50]. S ohledem na časté nadbytečné léčení PMM2-CDG pacientů substitucí levothyroxinem jen na základě vyšší hladiny TSH, se doporučuje sledovat výhradně parametry FT4 – hormonu, který není glykosylován. U dvou našich pacientů se rozvinula centrální hypothyreóza v rámci panhypopituitarismu, což zatím nebylo v literatuře popsáno.
Hypoglykémie v novorozeneckém a kojeneckém věku jsme zjistili u čtyř pacientů s PMM2-CDG. U dvou pacientů se jednalo o hypoglykémie v rámci deficitu ACTH a kortizolu, u jednoho chlapce v rámci jaterního selhání a u čtvrtého pacienta při multiviscerálním postižení s komplexní symp-tomatologií. Hladina inzulinu nebyla u těchto pacientů stanovena. U popsaných 24 novorozenců a kojenců s PMM2--CDG s opakovanými hypoglykémiemi v literatuře byl u 40 % z nich popsán hyperinzulinismus s dobrou odpovědí na diazoxid. Všechny adolescentní a dospělé dívky z našeho souboru mají dokumentovanou opožděnou pubertu v rámci hypergonadotropního hypogonadismu. Jen 3 dívky z 85 žen s PMM2-CDG s dokumentovanými gonadálními funkcemi měly normální nástup puberty a menarché [24, 45, 51]. Patofyziologie ovariálního selhání není přesně známa. Abnormální glykosylace gonadotropinů, jejich receptorů a pohlavních hormonů může vyústit v hypergonadotropní hypogonadismus, ale FSH in vitro měřená aktivita nebyla významně snížena [48]. U třech vyšetřovaných chlapců v adolescentním věku jsme zaznamenali normální pohlavní vývoj, i když v literatuře jsou popsané ojedinělé případy testikulární atrofie a hypogonadismu u mužů [40, 48, 52]. Snížená kostní denzita, v rozsahu osteopenie až po osteoporózu se začátkem již v dětství, je nalezena až u 60 % PMM2-CDG pacientů [30] a vede ke zvýšenému riziku fraktur (až ve 26 % případů) [18]. Denzitometrie prokázala u všech našich devíti vyšetřených pacientů sníženou kostní denzitu, dosahující u jedné pacientky v oblasti bederní páteře až hodnoty Z-skóre -6,8.
Koagulopatie
Charakteristickou laboratorní patologií u pacientů s PMM2-CDG je smíšená koagulopatie. Je přítomná u všech našich pacientů, s klinickým dopadem u přibližně jedné třetiny z nich. Postižené jsou jak prokoagulační, tak antikoagulační faktory. Nejčastější a nejvýznamnější patologie je popisována pro koagulační faktor IX, XI, antitrombin, protein C a S. V rámci poddiagnostikovanosti PMM2-CDG pacientů je nutné zdůraznit, že někteří pacienti s mírným fenotypem mohou být sledováni právě pro izolovaný deficit antitrombinu [53]. Trombotické příhody arteriální i venózní jsou popsány u 12,5 % pacientů, u čtyř pacientů došlo k trombóze mozkových žil, mezi závažné rizikové faktory trombóz patří operační zákrok a katetrizace [54]. Zajímavým nálezem je fluktuace tíže koagulační poruchy s věkem [55] a horečkou, pravděpodobně kvůli termolabilitě PMM2 [24, 56].
Diagnostika
Jako screeningovou metodu v diagnostice jsme používali IEF sérového transferinu, která je rychlá a senzitivní, ale nespecifická metoda k průkazu nemoci. Je založena na separaci transferinových isoforem na základě odlišného náboje vyplývajícího z různého počtu přítomných negativně nabitých terminálních zbytků sialové kyseliny. U PMM2-CDG obecně, jako i u všech našich pacientů v souboru, typicky nacházíme profil typu I se sníženým množstvím tetrasialovaných forem transferinu a zvýšením di - a asialovaných forem transferinu (obr. 4). V našem souboru jsme potvrdili poruchu PMM2 (10–30 % reziduální aktivity) u 84 % testovaných vzorků, pouze u dvou vzorků lymfocytů (P13 a P20) byla reziduální aktivita hraničně patologická na 52–63 % a u dvou vzorků fibroblastů 32–41 % (P7, P9) průměru kontrol. Až u 18 % publikovaných pacientů s PMM2-CDG byla v izolovaných lymfocytech a/nebo kultivovaných fibroblastech nalezena normální aktivita PMM2 [43, 57], proto nelze na enzymologické vyšetření spoléhat z důvodu rizika falešně negativního výsledku a vždy je nutno provést i molekulárně-genetické vyšetření.

Molekulárně-genetické vyšetření genu PMM2 je k dispozici na našem pracovišti. Gen PMM2 je lokalizován na chromosomu 16p13.2, má osm exonů a jeho proteinový produkt je o velikosti 246 aminokyselin. V genu pro PMM2 bylo nalezeno již 129 mutací, zahrnujících >95 bodových mutací, několik nonsense mutací, 8 mutací měnící čtecí rámec, >10 sestřihových mutací a jedna delece celého exonu 8 o velikosti 28 kb [58]. V našem souboru pacientů bylo zastoupeno 10 mutací v PMM2 (tab. 2), všichni pacienti jsou složení heterozygoti. Mutaci c.422G>A (p.Arg141His), která je nejpočetnější a je v literatuře popisována u 60 % pacientů, jsme nalezli i v našem souboru u 18 pacientů. Mutace c.422G>A nebyla dosud nalezena v homozygotní formě, a proto se předpokládá, že je v homozygotní formě neslučitelná se životem [59–61]. Mutace c.338C>T (p.Phe119Leu) byla nalezená u 13 našich subjektů, ve složené heterozygotní formě s mutací c.422G>A u 10 z nich. Obě mutace jsou tedy v České republice prevalentní.
ZÁVĚR
Domníváme se, že PMM2-CDG je v České republice, i 38 let od svého objevení, diagnózou málo známou a často opomíjenou. Důkazem pro to může být rozpor mezi odhadovanou prevalencí 1 : 20 000 [62] a tím, že na našem území bylo za 16 let od zavedení diagnostiky potvrzeno jen 22 pa-cientů s touto nemocí. Dále také mezi našimi pacienty nenacházíme pacienty s normálním či hraničním intelektem a mírnou neurologickou symptomatologií, jak je popsáno v literatuře [19–22]. Příčinou může být fakt, že zatímco dosud tuto diagnózu zvažujeme především v diferenciální diagnostice výrazné psychomotorické retardace a mozečkové atrofie, jedná se zřejmě o onemocnění s mnohem širším spektrem příznaků a heterogenní tíží fenotypu.
Věříme, že tato práce přispěje k lepšímu popisu přirozeného průběhu PMM2-CDG. Tato znalost je pro nás důležitá kvůli urychlení diagnostiky a možnostem co nejpřesnější informovanosti pacientů a jejich rodin. Dosud nebylo publikováno mnoho prací charakterizujících dlouhodobý vývoj pacientů s PMM2-CDG a jeho projevy v dospělosti. Proto je v plánu zařazení našich pacientů do prospektivní multicentrické mezinárodní studie hodnotící přirozený průběh onemocnění a eventuálně zařazení do klinické studie s novou léčebnou molekulou LipoM1P, která by měla zajistit dostupnost produktu narušené biochemické reakce přímo v buňkách.
Zkratky:
ALS (acid-labile subunit) – kyselá labilní podjednotka
aPTT – aktivovaný parciální tromboplastinový čas
CDG (congenital disorder of glycosylation) – dědičné poruchy glykosylace
CMP – cévní mozková příhoda
DOLK-CDG (dolichol kinase deficiency) – deficit dolicholkinázy
EMG – elektromyografie
GPI – glykosylfosfatidylinositol
IGF-1 (insulin-like growth factor 1) – inzulinu podobný růstový faktor
IGFBP3 (IGF binding protein 3) – IGF vázající protein 3
INR (international normalized ratio) – mezinárodní normalizovaný poměr
LipoM1P – manóza-1-fosfát inkorporovaná do liposomu
LLO (lipid-linked oligosaccharide) – oligosacharid vázaný na lipid
M1P – manóza-1-fosfát
MPI-CDG (mannosophosphate isomerase deficiency) – deficit fosfomanoisomerázy
NADP+, NADPH – nikotinamidadenindinukleotidfosfát a jeho redukovaná forma
PGM1-CDG (phosphoglucomutase deficiency type 1) – deficit fosfoglukomutázy 1
PMM2 – fosfomanomutáza 2
PMR – psychomotorická retardace
SLE (stroke-like episodes) – iktu podobné příhody
TBG (thyroxine-binding globulin) – tyroxin vázající globulin
IEF TRF (isoelectric focusing of transferrin) – isoelektrická fokusace transferinu
Podpořeno: MZ ČR – RVO VFN 64165, MZ ČR – AZV 16-31932A.
Korespondující autor:
Doc. MUDr. Tomáš Honzík, Ph.D.
Klinika dětského a dorostového lékařství
1. LF UK a VFN
Ke Karlovu 2
128 08 Praha 2
e-mail: tomas.honzik@vfn.cz
Sources
1. Corfield A, Berry M. Current aspects of eukaryotic glycosylation. Trends Biochem Sci 2015; 40 : 351–359.
2. Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta (BBA) – General Subjects 1999; 1473 (1): 4–8.
3. Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 2012; 13 (7): 448–462.
4. Jaeken J, Vanderschueren-Lodeweyckx M, Casaer P, et al. Familial psychomotor retardation with markedly fluctuating serum prolactin, FSH and GH levels, partial TBG-deficiency, increased serum arylsulphatase A and increased CSF protein: a new syndrome? Pediatr Res 1980; 14 : 179.
5. Ng BG, Freeze HH. Perspectives on glycosylation and its congenital disorders. Trends Genet 2018.
6. Hennet T, Cabalzar J. Congenital disorders of glycosylation: a concise chart of glycocalyx dysfunction. Trends Biochem Sci 2015; 40 (7): 377–384.
7. Jaeken J, Van Eijk H, Van der Heul C, et al. Sialic acid-deficient serum and cerebrospinal fluid transferrin in a newly recognized genetic syndrome. Clin Chim Acta 1984; 144 (2–3): 245–247.
8. Wopereis S, Grünewald S, Huijben KM, et al. Transferrin and apolipoprotein C-III isofocusing are complementary in the diagnosis of N - and O-glycan biosynthesis defects. Clin Chem 2007; 53 (2):1 80–187.
9. Lacey JM, Bergen HR, Magera MJ, et al. Rapid determination of transferrin isoforms by immunoaffinity liquid chromatography and electrospray mass spectrometry. Clin Chem 2001; 47 (3): 513–518.
10. Aebi M, Helenius A, Schenk B, et al. Carbohydrate-deficient glycoprotein syndromes become congenital disorders of glycosylation: An updated nomenclature for CDG. Glycoconj J 1999; 16 (11): 669–671.
11. Jaeken J, Hennet T, Matthijs G, et al. CDG nomenclature: time for a change! Biochim Biophys Acta – Mol Basis of Dis 2009; 1792 (9): 825–826.
12. Thiel C, Körner C. Therapies and therapeutic approaches in congenital disorders of glycosylation. Glycoconj J 2013; 30 (1): 77–84.
13. Grünewald S. The clinical spectrum of phosphomannomutase 2 deficiency (CDG-Ia). Biochim Biophys Acta – Mol Basis of Dis 2009; 1792 (9): 827–834.
14. Hagberg BA, Blennow G, Kristiansson B, et al. Carbohydrate-deficient glycoprotein syndromes: peculiar group of new disorders. Pediatr Neurol 1993; 9 (4): 255–262.
15. de Lonlay P, Seta N, Barrot S, et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet 2001; 38 (1): 14–19.
16. van de Kamp JM, Lefeber DJ, Ruijter GJ, et al. Congenital disorder of glycosylation type Ia presenting with hydrops fetalis. J Med Genet 2007; 44 (4): 277–280.
17. Wolthuis D, van Asbeck E, Kozicz T, et al. Abnormal fat distribution in PMM2-CDG. Mol Genet Metab 2013; 110 (3): 411–413.
18. Kjaergaard S, Schwartz M, Skovby F. Congenital disorder of glycosylation type Ia (CDG-Ia): phenotypic spectrum of the R141H/F119L genotype. Arch Dis Child 2001; 85 (3): 236–239.
19. Vuilleumier-Barrot S, Isidor B, Dupré T, et al. Expanding the spectrum of PMM2-CDG phenotype. In: JIMD Reports – Case and Research Reports. Vol. 5, edn.: Springer, 2011 : 123–125.
20. Giurgea I, Michel A, Le Merrer M, et al. Underdiagnosis of mild congenital disorders of glycosylation type Ia. Pediatr Neurol 2005; 32 (2): 121–123.
21. Pancho C, Garcia-Cazorla A, Varea V, et al. Congenital disorder of glycosylation type Ia revealed by hypertransaminasemia and failure to thrive in a young boy with normal neurodevelopment. J Pediatr Gastroenterol Nutr 2005; 40 (2): 230–232.
22. Shanti B, Silink M, Bhattacharya K, et al. Congenital disorder of glycosylation type Ia: Heterogeneity in the clinical presentation from multivisceral failure to hyperinsulinaemic hypoglycaemia as leading symptoms in three infants with phosphomannomutase deficiency. J Inherit Metab Dis 2009; 32 (1): 241–251.
23. Damen G, de Klerk H, Huijmans J, et al. Gastrointestinal and other clinical manifestations in 17 children with congenital disorders of glycosylation type Ia, Ib, and Ic. J Pediatr Gastroenterol Nutr 2004; 38 (3): 282–287.
24. Schiff M, Roda C, Monin M-L, et al. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J Med Genet 2017; 54 (12): 843–851.
25. Antoun H, Villeneuve N, Gelot A, et al. Cerebellar atrophy: an important feature of carbohydrate deficient glycoprotein syndrome type 1. Pediatr Radiol 1999; 29 (3): 194–198.
26. Neumann LM, von Moers A, Kunze J, et al. Congenital disorder of glycosylation type 1a in a macrosomic 16-month-old boy with an atypical phenotype and homozygosity of the N216I mutation. Eur J Pediatr 2003; 162 (10): 710–713.
27. Funke S, Gardeitchik T, Kouwenberg D, et al. Perinatal and early infantile symptoms in congenital disorders of glycosylation. Am J Med Genet Part A 2013; 161 (3): 578–584.
28. Barone R, Carrozzi M, Parini R, et al. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015; 262 (1):154–164.
29. Izquierdo-Serra M, Martinez-Monseny AF, Lopez L, et al. Stroke-like episodes and cerebellar syndrome in phosphomannomutase deficiency (PMM2-CDG): evidence for hypoglycosylation-driven channelopathy. Int J Mol Sci 2018; 19 (2): 619.
30. Monin M-L, Mignot C, De Lonlay P, et al. 29 French adult patients with PMM2-congenital disorder of glycosylation: outcome of the classical pediatric phenotype and depiction of a late-onset phenotype. Orphanet J Rare Dis 2014; 9 (1): 207.
31. Edwards M, McKenzie F, O‘callaghan S, et al. Prenatal diagnosis of congenital disorder of glycosylation type Ia (CDG–Ia) by cordocentesis and transferrin isoelectric focussing of serum of a 27–week fetus with non–immune hydrops. Prenat Diagn 2006; 26 (10): 985–988.
32. de Diego V, Martínez-Monseny AF, Muchart J, et al. Longitudinal volumetric and 2D assessment of cerebellar atrophy in a large cohort of children with phosphomannomutase deficiency (PMM2-CDG). J Inherit Metab Dis 2017; 40(5):709–713.
33. Hutchesson A, Gray R, Spencer D, et al. Carbohydrate deficient glycoprotein syndrome; multiple abnormalities and diagnostic delay. Arch Dis Child 1995; 72 (5): 445–446.
34. van der Knaap MS, Wevers RA, Monnens L, et al. Congenital nephrotic syndrome: a novel phenotype of type I carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1996; 19 (6): 787–791.
35. Coman D, McGill J, MacDonald R, et al. Congenital disorder of glycosylation type 1a: three siblings with a mild neurological phenotype. J Clin Neurosci 2007; 14 (7): 668–672.
36. Jamroz E, Adamek D, Paprocka J, et al. CDG type Ia and congenital cytomegalovirus infection: two coexisting conditions. J Child Neurol 2009; 24 (1): 13–18.
37. Silva MG, De Castro J, Stibler H, et al. Prenatal hypertrophic cardiomyopathy and pericardial effusion in carbohydrate-deficient glycoprotein syndrome. J Inherit Metab Dis 1996; 19 (2): 257–259.
38. Marquardt T, Hülskamp G, Gehrmann J, et al. Severe transient myocardial ischaemia caused by hypertrophic cardiomyopathy in a patient with congenital disorder of glycosylation type Ia. Eur J Pediatr 2002; 161 (10): 524–527.
39. Aronica E, van Kempen A, Van der Heide M, et al. Congenital disorder of glycosylation type Ia: a clinicopathological report of a newborn infant with cerebellar pathology. Acta Neuropathol 2005; 109 (4): 433–442.
40. Schoffer KL, O‘sullivan JD, McGill J. Congenital disorder of glycosylation type Ia presenting as early–onset cerebellar ataxia in an adult. Mov Disord 2006; 21 (6): 869–872.
41. Wurm D, Hänsgen A, Kim Y-J, et al. Early fatal course in siblings with CDG-Ia (caused by two novel mutations in the PMM2 gene): clinical, molecular and autopsy findings. Eur J Pediatr 2007; 166 (4): 377–378.
42. Ong BB, Gole GA, Robertson T, et al. Retinal hemorrhages associated with meningitis in a child with a congenital disorder of glycosylation. Forensic Sci Med Pathol 2009; 5 (4): 307–312.
43. Grünewald S, Schollen E, Van Schaftingen E, et al. High residual activity of PMM2 in patients’ fibroblasts: possible pitfall in the diagnosis of CDG-Ia (phosphomannomutase deficiency). Am J Hum Genet 2001; 68 (2): 347–354.
44. Arnoux J, Boddaert N, Valayannopoulos V, et al. Risk assessment of acute vascular events in congenital disorder of glycosylation type Ia. Mol Genet Metab 2008; 93 (4): 444–449.
45. Perez-Duenas B, García-Cazorla A, Pineda M, et al. Long-term evolution of eight Spanish patients with CDG type Ia: typical and atypical manifestations. Eur J Paediatr Neurol 2009; 13 (5): 444-451.
46. Enns GM, Steiner RD, Buist N, et al. Clinical and molecular features of congenital disorder of glycosylation in patients with type 1 sialotransferrin pattern and diverse ethnic origins. J Pediatr 2002; 141 (5): 695–700.
47. Grünewald S, De Vos R, Jaeken J. Abnormal lysosomal inclusions in liver hepatocytes but not in fibroblasts in congenital disorders of glycosylation (CDG). J Inherit Metab Dis 2003; 26 (1): 49–54.
48. de Zegher F, Jaeken J. Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 1995; 37 (4 Pt 1): 395–401.
49. Miller BS, Duffy MM, Addo OY, et al. rhIGF-1 therapy for growth failure and IGF-1 deficiency in congenital disorder of glycosylation Ia (PMM2 deficiency). J Investig Med High Impact Case Rep 2013 Sep 5; 1 (3): 2324709613503316.
50. Jaeken J, Hagberg B, Strømme P. Clinical presentation and natural course of the carbohydrate–deficient glycoprotein syndrome. Acta Pædiatrica 1991; 80 : 6–13.
51. Pineda M, Pavia C, Vilaseca M, et al. Normal pubertal development in a female with carbohydrate deficient glycoprotein syndrome. Arch Dis Child 1996; 74 (3): 242.
52. Krasnewich D, O‘Brien K, Sparks S. Clinical features in adults with congenital disorders of glycosylation type Ia (CDG–Ia). Am J Med Genet Part C Semin Med Genet 2007; 145c (3): 302–306.
53. de la Morena-Barrio ME, Hernández-Caselles T, Corral J, et al. GPI-anchor and GPI-anchored protein expression in PMM2-CDG patients. Orphanet J Rare Dis 2013; 8 (1): 170.
54. Linssen M, Mohamed M, Wevers R, et al. Thrombotic complications in patients with PMM2-CDG. Mol Genet Metab 2013; 109 (1): 107–111.
55. Ono H, Sakura N, Yamashita K, et al. Novel nonsense mutation (R194X) in the PMM2 gene in a Japanese patient with congenital disorder of glycosylation type Ia. Brain Dev 2003; 25 (7): 525–528.
56. Le Bizec C, Vuillaumier–Barrot S, Barnier A, et al. A new insight into PMM2 mutations in the French population. Hum Mutat 2005; 25 (5): 504–505.
57. Westphal V, Peterson S, Patterson M, et al. Functional significance of PMM2 mutations in mildly affected patients with congenital disorders of glycosylation Ia. Genet in Medicine 2001; 3 (6): 393.
58. Schollen E, Keldermans L, Foulquier F, et al. Characterization of two unusual truncating PMM2 mutations in two CDG-Ia patients. Molec Genet Metab 2007; 90 (4): 408–413.
59. Kjaergaard S, Skovby F, Schwartz M. Absence of homozygosity for predominant mutations in PMM2 in Danish patients with carbohydrate-deficient glycoprotein syndrome type 1. Eur J Hum Genet 1998; 6 (4): 331.
60. Matthijs G, Schollen E, Van Schaftingen E, et al. Lack of homozygotes for the most frequent disease allele in carbohydrate-deficient glycoprotein syndrome type 1A. Am J Hum Genet 1998; 62 (3): 542–550.
61. Pirard M, Matthijs G, Heykants L, et al. Effect of mutations found in carbohydrate-deficient glycoprotein syndrome type IA on the activity of phosphomannomutase 2. FEBS Letters 1999; 452 (3): 319–322.
62. Schollen E, Kjaergaard S, Legius E, et al. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia). Eur J Hum Genet 2000; 8 (5): 367.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2018 Issue 6
Most read in this issue
- Charakteristické klinické příznaky a laboratorní odchylky dědičných poruch metabolismu
- Komplexný pohľad na deficit vitamínu B12 v detskom veku
- Novorozenecký screening dědičných metabolických poruch v České republice
- Lyzozómové choroby – vývoj diagnostiky a liečby na Slovensku