
Specifické a asociované autoprotilátky u polymyozitidy a dermatomyozitidy
Authors:
M. Modrá; J. Vencovský
Authors‘ workplace:
Revmatologický ústav, Praha
Published in:
Čes. Revmatol., 16, 2008, No. 4, p. 169-176.
Category:
Overview Reports
Overview
Polymyozitida a dermatomyozitida patří mezi idiopatické zánětlivé myopatie. Je pro ně charakteristická řada klinických příznaků a v určení přesné diagnózy může pomoci i protilátkový profil nemocného. Autoprotilátky u polymyozitidy a dermatomyozitidy dělíme do několika skupin: pro myozitidu specifické protilátky, s myozitidou asociované protilátky a tkáňově specifické protilátky. Nejdůležitější jsou specifické protilátky, které mají vysokou vypovídající hodnotu při stanovování diagnózy. V článku je podán základní přehled autoprotilátek, které se nacházejí u myozitid. Jsou zmíněny i nově objevené autoprotilátky, jako jsou anti-CADM-140, anti-p140/155 a anti-SAE.
Klíčová slova:
polymyozitida, dermatomyozitida, autoprotilátky
Úvod
Idiopatické zánětlivé myopatie (IZM) jsou heterogenní skupinou autoimunitních onemocnění, které jsou charakterizovány svalovou slabostí, elektromyografickými abnormalitami, zvýšenými hladinami enzymů svalového původu, zánětlivým infiltrátem ve svalech a přítomností autoprotilátek v krvi. Pro základní rozdělení se vymezují tři hlavní skupiny: primární idiopatická polymyozitida (PM), primární idiopatická dermatomyozitida (DM) a myozitida s inkluzními tělísky (IBM) (1). Diagnostika těchto onemocnění není v řadě případů jednoduchá, a pokud je přítomna správně stanovená autoprotilátka, je významnou pomocí v procesu určení nemoci. Jen ojediněle se ale zdá, že určení hladiny autoprotilátky může pomoci v ohodnocení aktivity onemocnění.
Autoprotilátky přítomné v krvi nemocných s myozitidou se dělí do tří skupin: autoprotilátky specifické pro myozitidu (MSA), autoprotilátky asociované s myozitidou (MAA) a tkáňově specifické autoprotilátky. MSA jsou pro myozitidu vysoce specifické a mají velkou vypovídací hodnotu, MAA a tkáňově specifické autoprotilátky se vyskytují jak u myozitid, tak i u jiných autoimunitních onemocnění (2).
Autoprotilátky specifické pro myozitidu
V mnoha studiích se prokázalo, že MSA jsou asociovány se specifickými klinickými projevy a mohou vést k pochopení patofyziologie idiopatických zánětlivých myopatií. Vyskytují u 40–50 % pacientů s myozitidou a dělíme je na podkladě antigenu, proti kterému jsou protilátky namířeny. Mezi nejvýznamnější z nich patří protilátky proti aminoacyl-tRNA syntetázám, proti cytoplazmatickému proteinu SRP (signal recognition particle) a proti nukleozomovému remodelačnímu komplexu Mi-2.
V naprosté většině případů bývá detekována pouze jedna MSA, nicméně existují i takové případy kdy byly detekovány dvě tyto protilátky současně. Například Ghirardello a kol. detekovali současně u jednoho pacienta anti-Jo-1 a anti-Mi-2 (3) a v pozdější studii ještě anti-Jo-1 spolu s anti-SRP (4).
Anti-aminoacyl-tRNA syntetázy
Aminocyl-tRNA syntetázy jsou cytoplazmatické enzymy, které katalyzují vazbu aminokyseliny na její příslušnou transferovou ribonukleovou kyselinu (tRNA). Hrají zcela zásadní roli v syntéze proteinů (5). Protilátky proti aminoacyl-tRNA-syntetázám se vyskytují především u DM a PM, ale mohou být výjimečně detekovány i u IBM. Přítomnost těchto protilátek je nejčastěji asociována s tzv. antisyntetázovým syndromem který je charakterizován vedle myozitidy přítomností intersticiální plicní fibrózy (ILD), poměrně těžké artritidy, Raynaudova fenoménu, prstů mechanika a horečnatých stavů (6, 7).
Anti-Jo-1
Nejčastěji detekovanou autoprotilátkou u pacientů s myozitidou je anti-Jo-1, antisyntetázová protilátka namířená proti histidyl-tRNA syntetáze. Tato protilátka je pro toto onemocnění diagnostickým ukazatelem. Její specificita se blíží 100 %, senzitivita se pohybuje mezi 24 až 30 % (18–46 % pro PM) u dospělých. U juvenilních forem myozitidy bývá anti-Jo-1 detekována zřídka. Pacienti pozitivní na anti-Jo-1 protilátky mají zpravidla závažné klinické projevy a horší prognózu. Více než 70 % z nich trpí intersticiální plicní fibrózou, její přítomnost je také hlavním rozdílem mezi anti-Jo-1 negativními a pozitivními pacienty (9). Stone a kol. se zabývali vztahy mezi hladinami anti-Jo-1 protilátek a aktivitou. Vyšetřili 81 pacientů a nalezli korelaci mezi hladinou anti-Jo-1, hladinou kreatinkinázy v séru, aktivitou nemoci hodnocenou pomocí nástroje MITAX a postižením kloubů (10, 11).
Anti-PL-7
Anti-PL-7 je antisyntetázová protilátka nasměrovaná proti threonyl-tRNA synthetáze (8). Bývá obvykle detekována u 5–10 % pacientů s myozitidou (9). Je, stejně jako anti-Jo-1, asociována s intersticiální plicní fibrózou. Svalové postižení bývá u anti-PL-7 pozitivních pacientů o něco mírnější (12).
Anti-PL-12
Anti-PL-12 je antisyntetázová protilátka proti alanyl-tRNA synthetáze. Tato protilátka je asociována s podskupinou nemocných, u kterých je vysoká frekvence ILD a artritidy, stejně jako je tomu i u anti-Jo-1 pozitivních pacientů (13).
Anti-EJ
Anti-EJ je antisyntetázová protilátka proti glycyl-tRNA synthetáze. Tato protilátka bývá spojována s pacienty, kteří mají typickou dermatomyozitickou vyrážku a ILD. Může být u pacienta detekována ještě před vzplanutím choroby. U pacientů anti-EJ pozitivních byl též pozorován Raynaudův fenomén a ostatní klinické projevy typické pro antisyntetázový syndrom (14).
Anti-KS (Anti-AsnRS)
Anti-KS je antisyntetázová protilátka proti asparaginyl-tRNA synthetáze. Hirakata a kol. při vyšetřování 2500 pacientů se zánětlivým onemocněním pojiva detekovali anti-KS protilátku u 8 pacientů, 2 z nich měli DM, 7 mělo ILD, 4 artritidu a 1 Raynaudův fenomén (5). Toto zjištění je v souladu s Hirakatovou předchozí studií, kde anti-KS protilátky detekovali u dvou pacientů s ILD, z nichž ani jeden neměl myozitidu. Tudíž vše nasvědčuje tomu, že anti-KS protilátka je asociována víc s ILD než se samotnou myozitidou (15).
Anti-OJ
Anti-OJ je antisyntetázová protilátka proti isoleucyl-tRNA syntetáze. Sato a kol. vyšetřili 1135 japonských pacientů s autoimunitním onemocněním a 48 zdravých kontrol na anti-OJ protilátky. Anti-OJ detekovali u 3 pacientů s PM a u jednoho s překryvným syndromem PM/RA. Všichni tito pacienti měli ILD, Raynaudův fenomén a sklerodaktylii. To nasvědčuje tomu, že anti-OJ autoprotilátka je charakteristická pro podtyp antisyntetázového syndromu, pro který je typičtější ILD než myozitida a Raynaudův fenomén (16).
Anti-Zo
Anti-Zo protilátka je zatím poslední popsanou antisyntetázovou autoprotilátkou. Jde o protilátku namířenou proti fenylalanyl-tRNA synthetáze. Byla popsaná skupinou z Bathu, kde pomocí proteinové imunoprecipitace detekovali v séru pacienta s myozitidou přítomnost dvou proteinů o molekulové hmotnosti 60 a 70 kDa a odhalili, že pozitivní proužky odpovídají tRNA syntetáze (17).
Anti-SRP
Anti-signal recognition particle jsou protilátky proti 325 kDa velkému ribonukleoproteinu složenému ze 7 SL RNA a šesti peptidů: 9, 14, 19, 54, 68 a 72 kDa asociovaných s ribozomy. SRP komplex se účastní transportu proteinů do endoplazmatického retikula během proteosyntézy. Většina studií ukazuje, že anti-SRP autoprotilátky jsou nejčastěji detekovány u PM, někdy jsou ale přítomné i u DM a IBM. Hengstmanova studie potvrzuje, že přítomnost anti-SRP protilátek se pojí s relativně agresivním klinickým obrazem, charakterizovaným rychlým začátkem onemocnění, těžkým průběhem s myalgiemi a artralgiemi, vysokými hladinami kreatinkinázy v séru a často špatnou odpovědí na imunosupresivní léky (18). Předpokládaná asociace mezi srdečním postižením a anti-SRP protilátkami je založena na dvou studiích s celkovým počtem 20 pacientů (7, 19). Ale existují i studie, kde častější přítomnost srdečního postižení u anti-SRP pozitivních pacientů nebyla prokázána (18,20). Miller a kol. potvrdili, že anti-SRP autoprotilátky vymezují specifickou autoimunitní myopatii charakterizovanou rychle progredující svalovou slabostí, nedostatečnou odpovědí na glukokortikoidy a žádnými klinickými známkami multiorgánového postižení (21). Histologicky jsou nejčastějším nálezem nekrotické změny svalových vláken při minimálním zánětlivém infiltrátu nebo při jeho úplné absenci (20).
Sezonní změny v nástupu a progresi nemoci byly pozorovány u řady autoimunitních onemocnění, včetně PM/DM. Leff a kol. popsali, že u anti-SRP pozitivních pacientů dochází k rozvoji myozitidy nejčastěji v období mezi zářím a únorem (22). Tento trend sezonního nástupu nemoci potvrzuje i Millerova studie (21).
Anti-Mi-2
Anti-Mi-2 je autoprotilátka proti proteinovému komplexu (složenému z proteinů o velikosti: 240, 200, 150, 72, 65, 64, 50 a 40 kDa), který se účastní transkripce. Tyto proteiny tvoří centrální komponentu takzvaného nukleozomového remodelačního a histon deacetylačního komplexu (NuRD), který se účastní při regulaci transkripce pomocí deacetylce histonů (23). Anti-Mi-2 protilátka byla nazvána podle její přítomnosti v séru 60leté pacientky s dermatomyozitidou (Pacientka Mi). Anti-Mi-2 je jedinou dobře popsanou MSA proti jadernému antigenu. Molekulární struktura Mi-2 antigenu byla v roce 1995 studována dvěma výzkumnými skupinami (24, 25). Obě identifikovali protein, který reagoval s anti-Mi-2 pozitivním sérem. Proteiny sice nebyly totožné, nicméně byl prokázán vysoký stupeň podobnosti. Byly tudíž pojmenovány Mi-2α a Mi-2β (26).
Anti-Mi-2 protilátky jsou diagnostickým markrem pro idiopatické zánětlivé myozitidy se senzitivitou mezi 4 až 18 %. Bývají detekovány u 15–31 % pacientů s dermatomyozitidou a u 10–15% pacientů s juvenilní dermatomyozitidou. Diagnostická specificita pro DM je 99 %. Téměř všichni pacienti, kteří jsou anti-Mi-2 pozitivní, mají dermatomyozitidu, pouze 3 % z nich mají polymyozitidu. Mi-2 protilátky bývají detekovány již v časných fázích onemocnění. Oproti pacientům, kteří jsou pozitivní na antisyntetázové protilátky, mají Mi-2 pozitivní pacienti relativně mírný klinický obraz, zřídka mívají synovitidu, plicní postižení nebo Raynaudův fenomén (9). Hengstman a kol. popsali, že anti-Mi-2 nejsou asociovány s žádným specifickým klinickým projevem, kromě dermatomyozitické vyrážky (27). Anti-Mi-2 pozitivní pacienti mívají obvykle dobrou prognózu spojenou s relativně nízkým rizikem rakoviny (28). O’Callaghan a kol. detekovali ve své studii 7,5 % anti-Mi-2 pozitivních pacientů. Všichni tito pacienti měli DM. Jejich studie také podpořila myšlenku, že anti-Mi-2 je charakteristická protilátka pro DM pacienty, kteří trpí kožními lézemi a dysfágií (29).
Anti-CADM-140
Sato a kol. detekovali anti-CADM-140 protilátky v 8 sérech z 298 pacientů se zánětlivým onemocněním pojiva a ILD. Všech těchto 8 pacientů mělo dermatomyozitidu bez klinických příznaků (clinical-amyopathic DM = C-ADM), žádný z nich neměl PM, klasickou DM nebo jiné zánětlivé onemocnění pojiva. Jejich séra reagovala imunoprecipitací a na imunoblotu s polypeptidem o přibližné velikosti 140 kDa. Imunofluorescence byla v těchto sérech detekována cytoplazmatická a zrnitá. Všichni tito anti-CADM pozitivní pacienti měli výrazně rychle progredující ILD. Zdá se, že přítomnost této protilátky by mohla být diagnostickým kritériem pro C-ADM (30).
Protilátka proti jadernému proteinu o molekulové hmotnosti 155 a 140 kDa
Kaji a kol. popsali novou autoprotilátku reagující se 155 a 140 kDa velkými jadernými proteiny. Detekovali tuto protilátku u 13 % japonských pacientů s DM. Tito všichni nemocní měli heliotropní vyrážku, Gottronovy papuly a erytém významně častěji než negativní pacienti. Žádný z těchto nemocných netrpěl intersticiální plicní fibrózou, ale významně častěji se u nich vyskytovalo nádorové bujení (31). Targoff a kol. potvrdili, že anti-p155 je protilátka charakteristická pro DM asociovanou s nádorovým bujením. Anti-p155 je jednou z nejčastěji detekovaných protilátek u této skupiny pacientů. Vyskytuje se stejně často u dospělých jako u dětí (32). Gunawardena a kol. vyšetřili 116 dětí trpících JDM. Detekovali zde anti-p155/140 protilátku s mnohem vyšší frekvencí u mužské části této skupiny. Pacienti anti-p155/140 měli signifikantně častěji kožní postižení včetně Gottronových papul, ulcerací a edémů (33). Chinoy a kol. zkoumali, zda by se pravděpodobnost rozvoje rakovinného bujení dala u pacientů s myozitidou nějak predikovat pomocí autoprotilátkového profilu. Asociace mezi myozitidou a zhoubným bujením není zcela objasněna, ale je známo, že riziko vzniku nádoru je o něco vyšší u DM než u PM. Bohužel zatím nebyly vyvinuty žádné spolehlivé metody pro predikci vzniku nádoru u těchto pacientů. Ve studii Chinoye bylo vyšetřeno 16 pacientů s nádorově asociovanou myozitidou (CAM). Tito pacienti měli pozdější nástup nemoci a byli negativní na rutinně testované myozitické protilátky (anti-Jo-1, anti-PM-Scl, anti-U1-RNP, anti-U3-RNP, anti-Ku). Přítomnost protilátek proti 155 kDa/140 kDa byla detekována pouze u DM a ukázala se jako významný faktor, který predikuje vznik CAM. Navrhují zavést vyšetření anti-p155/140 do rutinního testování (34).
Objev těchto protilátek má značný význam, neboť přináší nový pohled na klasifikaci zánětlivých idiopatických myopatií (35).
Anti-SAE
Jako antigenní cíl u této protilátky byl popsán lidský SUMO-1 (small ubiqutin-like modifier 1), což je 11 kDa velký protein, který má 18 % sekvenční homologii s ubiquitinem. Ubiquitin je malý protein, který je kovalentně vázán na proteiny určené k degradaci v proteasomu. Enzym SUMO-1 se aktivně podílí na posttranslačních modifikacích. Autoprotilátka byla identifikována u 2 ze 20 nemocných s DM (36). Oba tito pacienti měli těžší kožní onemocnění, dysfagii a ILD.
Anti-56 kDa
Anti-56 kDa protilátka je namířena proti jadernému ribonukleoproteinovému komplexu. Arad-Dann a kol. detekovali tyto protilátky v 85 % sér myozitických pacientů, častěji však u pacientů s počínající dermatomyozitidou (37). Zdálo se, že u dospělých pacientů přítomnost protilátky proti 56 kDa velkému proteinu koreluje s aktivitou onemocnění. Z ne zcela jasných důvodů není tato protilátka rutinně vyšetřována a to ani autory původního popisu.

S myozitidou asociované protilátky
Anti-PM-Scl
PM-Scl je intracelulární makromolekulární komplex skládající se z 11 až 16 proteinů (velkých od 20 do 110 kDa, včetně dvou hlavních peptidů, 100 a 75 kDa), lokalizovaných v jádře a cytoplazmě. Tyto proteiny mají převážně exoribonukleázovou aktivitu, PM-Scl-100 hraje hlavní roli během tvoření konce 5.8 S rRNA (38). Proteiny PM-Scl-100 a PM-Scl-75 jsou hlavními cílovými antigeny anti-PM-Scl autoprotilátek (39). Anti-PM-Scl protilátky jsou sérologickým markrem pro sklerodermii, polymyozitidu a především pro překryvný syndrom mezi těmito dvěma onemocněními. Anti-PM-Scl bývají detekovány přibližně u 24% pacientů s překryvným syndromem mezi sklerodermií a polymyozitidou a u 3–10 % pacientů se samotnou sklerodermií nebo polymyozitidou (40). Přibližně 50–70 % anti-PM-Scl pozitivních pacientů má PM/Scl překryvný syndrom a 20 % idiopatickou myozitidu (9).
Anti-Ku
Anti-Ku protilátky jsou namířeny proti heterodimeru (složenému z 80 a 70 kDa velkých proteinů) asociovanému s DNA-dependentní protein kinázou (350 kDa) (41). Anti-Ku protilátky se vyskytují u 5–25 % pacientů, kteří mají překryvný syndrom polymyozitidy se sklerodermií a u 1–7 % pacientů, kteří trpí myozitidou. Anti-Ku protilátka je typická pro pacienty, kteří mají svalové postižení a plicní komplikace, jak tomu bývá u překryvného syndromu (9). Frekvence přítomnosti anti-Ku protilátek se výrazně liší podle použitých metodik stanovení a jejich přesnou frekvenci bude teprve nutné stanovit (42, 43).
Anti-U1-snRNP
Anti-U1-snRNP jsou autoprotilátky proti malým jaderným ribonukleoproteinům: A (33 kDa), C (21 kDa) a 68 kDa (67–70 kDa). Vyskytují se nejčastěji u pacientů s překryvnými syndromy, především u smíšeného onemocnění pojiva (MCTD). Pokud jsou přítomny u SLE, bývá u těchto nemocných i myozitida (44). Pravděpodobně definují podskupinu myozitických pacientů charakterizovanou příznivějším průběhem onemocnění (45).
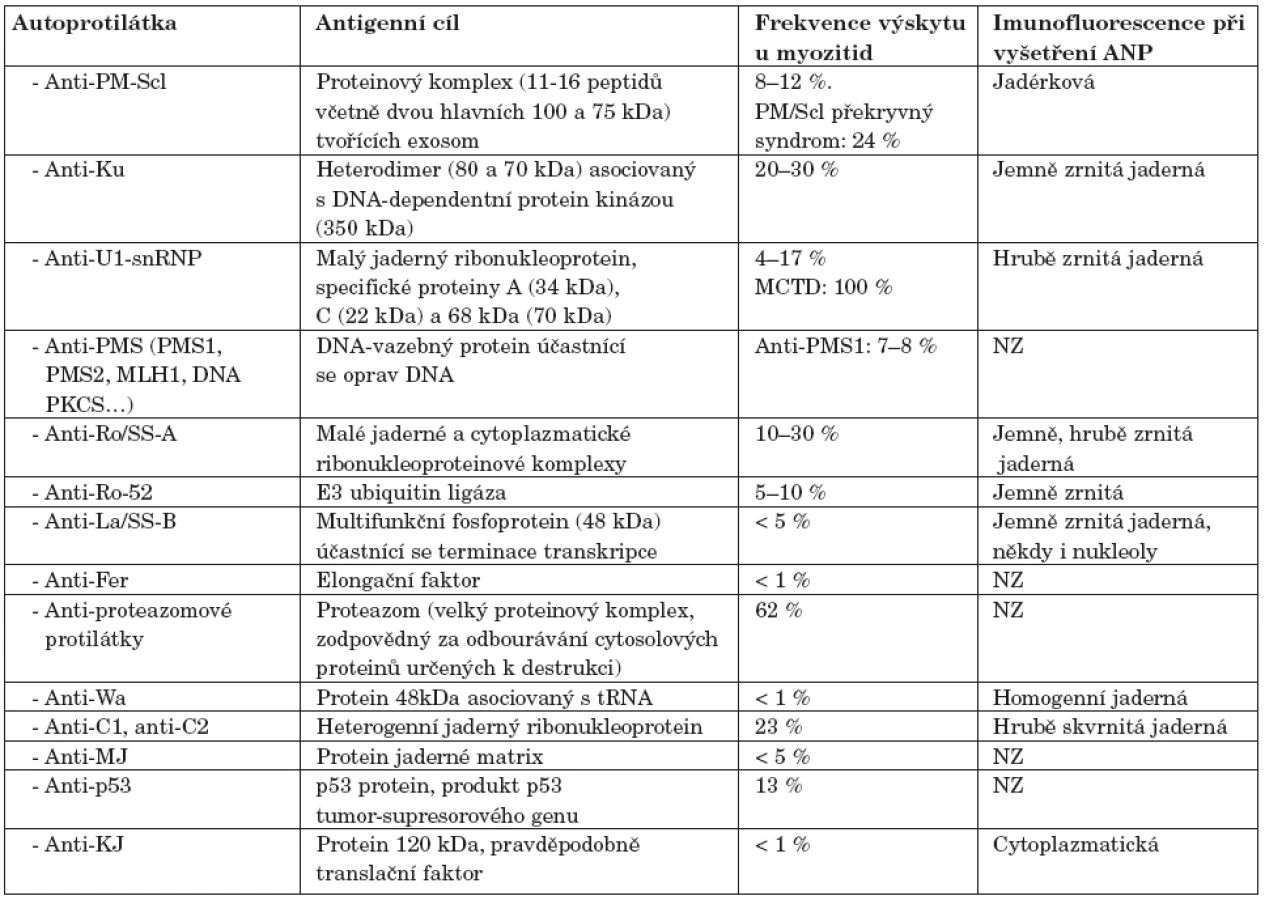
Protilátky proti proteazomu
Proteazom je proteinový komplex lokalizovaný v jádře a cytoplazmě, který se účastní, na ubiquitinu závislé, selektivní degradaci proteinů, při prezentaci antigenů molekulami HLA I. Protilátky proti proteazomu nejsou specifické pro jedno konkrétní onemocnění. Bývají detekovány u 30–60 % pacientů s autoimunitním systémovým onemocněním, nejčastěji (v 62 %) je nacházíme u myozitidy (46). Pacienti pozitivní na protilátky proti proteazomu bývají většinou zároveň anti-Jo-1 negativní (9).
Anti-Ro/SSA-A
Ro-52 a Ro-60 jsou malé jaderné a cytoplazmatické ribonukleoproteinové komplexy. Za antigen proti kterému je anti-Ro-52 protilátka namířena byla označena E3 ubiquitinová ligáza (47). Rutjes a kol. detekovali anti-Ro-52 protilátky ve 20 % ze 112 sér nemocných s idiopatickými zánětlivými myopatiemi. Popsali také, že protilátky anti-Ro-52 jsou asociovány s PM a DM a často se vyskytují (v 58 %) v sérech, které jsou též anti-Jo-1 pozitivní (48). Frank a kol. detekovali anti-Ro-52 u 58–70 % pacientů s autoimunitním onemocněním, kteří byli pozitivní na anti-aminoacyl-tRNA-syntetázové protilátky (v 69 % u pacientů s anti-PL-7, v 67 % s anti-PL-12, a v 70 % s anti-Jo-1), ve 43 % anti-SRP pozitivních pacientů a ve 47 % pacientů, kteří byli anti-PM-Scl pozitivní (49). O’Callaghan a kol. detekovali anti-Ro-52 protilátky u 20 % myozitických pacientů. Tito pacienti měli především antisyntetázový syndrom (29).
Anti-Ro-60 jsou často asociovány s protilátkami proti anti-Ro-52 a anti-La. Rutjes a kol. detekovali anti-Ro-60 a anti-La ve 4% sér s idiopatickou zánětlivou myozitidou (48). O’Callaghan a kol. detekovali anti-Ro-60 protilátky u 33% myozitických pacientů (29).
Anti-La/SS-B
Anti-La jsou autoprotilátky proti multifunkčnímu fosfoproteinu o velikosti 48 kDa, který se účastní terminace transkripce. Vytváří makromolekulární komplex spolu s antigenem Ro a malými, na uridin bohatými, molekulami RNA (hY1, hY3, hY4 a hY5 RNA). Tato protilátka je diagnostickým markerem především pro Sjögrenův syndrom (9) a u myozitid se vyskytuje zřídka.
Anti-Wa
Anti-Wa je antisytetázová protilátka namířená proti s tRNA asociovanému proteinu o molekulové hmotnosti 48kDa. Kajihara a kol. (46) popsali anti-Wa protilátku u dvou pacientů s myozitidou a intersticiální plicní fibrózou. Nicméně se ale zdá, že tato protilátka nebude mít nijak zvlášť velký klinický význam, neboť se vyskytuje pouze u velmi malého počtu nemocných a není pro myozitidu specifická. Výrazně častěji bývá detekována u pacientů se systémovou sklerodermií (50).
Anti-PMS1
Anti-PMS1 je protilátka proti DNA vazebnému proteinovému komplexu, který se účastní oprav a remodelace DNA. Casciola-Rosen a kol. (51) ve své studii detekovali tuto protilátku u 8 % pacientů s myozitidou.
Anti-C1, Anti-C2
Anti-C1 a C2 hnRNP byly poprvé identifikovány u pacienta se sklerodermií a psoriatickou artritidou (52). Heegaard a kol. (53) zjistili autoprotilátky proti C1 a C2 heterogenním jaderným ribonukleoproteinům u tří pacientů s myozitidou.
Anti-C1D
Schilders a kol. (54) prokázali, že nedávno popsaný, s exosomem asociovaný protein C1D, je významným autoantigenem u pacientů s PM/Scl překryvným syndromem. Autoprotilátky rozeznávající C1D byly detekovány u 7 z 30 pacientů (23 %) s překryvným syndromem mezi polymyozitidou a sklerodermií. Tato frekvence byla podobná frekvenci s jakou byly detekovány u této skupiny autoprotilátky proti PM-Scl-75 (27 %) a PM-Scl-100 (23 %). Protilátky proti C1D antigenu byly pozorovány pouze u dvou z 204 pacientů s dalšími autoimunitními chorobami, mezi kterými byli pacienti a PM, DM a sklerodermií.
Anti-MJ
Anti-MJ je protilátka směrovaná proti proteinu jaderné matrix. Její popis byl podán pouze formou abstraktu (55). Bývá výrazně častěji detekována u pacientů s juvenilní DM, než u jiných typů myozitid. Skupina anti-MJ pozitivních pacientů nemá žádné charakteristické shodné příznaky, ale asociace s juvenilní DM je velmi zajímavá, neboť pouze velmi malé množství těchto pacientů je pozitivní na některou jinou autoprotilátku.
Anti-p53
Mimura a kol. ve své studii prokázali, že anti-p53 protilátka, která bývá přítomna u pacientů s nádorovým bujením, se též vyskytuje u nemocných s PM/DM významně častěji než u zdravých lidí. Anti-p53 protilátku detekovali u 13 % (4/31) PM/DM pacientů. Jeden z těchto pacientů měl též zhoubný nádor (56).
Anti-KJ
U dvou pacientů s polymyozitidou, ILD a Raynaudovým fenoménem byla detekována protilátka, která silně inhibovala translaci mRNA. Tato autoprotilátka byla pojmenována anti-KJ. KJ antigen zřejmě nepatří mezi integrální ribozomální proteiny ani syntetázy, ale mohl by být translačním faktorem (57). Tato protilátka je detekována zcela výjimečně.
Shrnutí
Přestože naše znalosti biochemické povahy autoantigenů výrazně rostou, o patogenezi idiopatických zánětlivých myopatií toho stále ještě mnoho nevíme. Podobně jako u jiných autoimunitních onemocnění neznáme přesné důvody, proč buňky pacientů s myozitidou produkují specifické autoprotilátky. Přesto však autoprotilátky hrají významnou úlohu v diagnostice a klasifikaci onemocnění a pomáhají nám porozumět mechanismům odehrávajícím se v těle pacienta během průběhu nemoci. Specificita protilátek pro onemocnění, jejich vzájemná výlučnost a zjevné klinické asociace ukazují na to, že jejich tvorba má co do činění s etiologií onemocnění. Objevy dalších protilátek nám pomohou specifikovat další podskupiny myozitických onemocnění.
Tato studie byla podpořena výzkumnými záměry MSM 0021620812 Ministerstva školství, mládeže a tělovýchovy České republiky
Mgr. M. Modrá
Revmatologický ústav
Na Slupi 4
120 58 Praha 2
Sources
1. Dalakas MC. Polymyositis, dermatomyositis and inclusion body myositis. N Engl J Med 1991; 325 : 1478–98.
2. Al-Lozi M, Pestronk A. Organ-specific autoantibodies with muscle weakness. Curr Opin Rheumatol 1999; 11 : 483–8.
3. Ghirardello A, Zampieri S, Iaccarino L, Tarricone E, Tonello M, Bendo R, et al. Myositis specific and myositis associated autoantibodies in idiopathic inflammatory myopathies: a serologic study of 46 patients]. Reumatismo 2005; 57 : 22–8.
4. Ghirardello A, Zampieri S, Tarricone E, Iaccarino L, Bendo R, Briani C, et al. Clinical implications of autoantibody screening in patients with autoimmune myositis. Autoimmunity 2006; 39 : 217–21.
5. Hirakata M, Suwa A, Takada T, Sato S, Nagai S, Genth E, et al. Clinical and immunogenetic features of patients with autoantibodies to asparaginyl-transfer RNA synthetase. Arthritis Rheum 2007; 56 : 1295–303.
6. Matsushita T, Hasegawa M, Fujimoto M, Hamaguchi Y, Komura K, Hirano T, et al. Clinical evaluation of anti-aminoacyl tRNA synthetase antibodies in Japanese patients with dermatomyositis. J Rheumatol. 2007; 34 : 1012–8.
7. Love LA, Leff RL, Fraser DD, Targoff IN, Dalakas M, Plotz PH, Miller FW. A new approach to the classification of idiopathic inflammatory myopathy: myositis-specific autoantibodies define useful homogeneous patient groups. Medicine (Baltimore) 1991 : 70 : 360–74.
8. Targoff IN, Arnett FC, Reichlin M. Antibody to threonyl-transfer RNA synthetase in myositis sera. Arthritis Rheum 1988; 31 : 515–24.
9. Karsten C, Werner S, Falk H, Fritzer MJ. Autoantibodies in systemic autoimmune diseases-a diagnostic reference. Pabs Scientific publishers. Berlin. 2007. 2nd edition.
10. Stone KB, Oddis CV, Fertig N, Katsumata Y, Lucas M, Vogt M, et al. Anti-Jo-1 antibody levels correlate with disease activity in idiopathic inflammatory myopathy. Arthritis Rheum 2007; 56 : 3125–31.
11. Isenberg DA, Allen E, Farewell V, Ehrenstein MR, Hanna MG, Lundberg IE, et al. International consensus outcome measures for patients with idiopathic inflammatory myopathies. Development and initial validation of myositis activity and damage indices in patients with adult onset disease. Rheumatology (Oxford) 2004; 43 : 49–54.
12. Yamasaki Y, Yamada H, Nozaki T, Akaogi J, Nichols C, Lyons R, et al. Unusually high frequency of autoantibodies to PL-7 associated with milder muscle disease in Japanese patients with polymyositis/dermatomyositis. Arthritis Rheum 2006; 54 : 2004–9.
13. Targoff IN, Arnett FC. Clinical manifestations in patients with antibody to PL-12 antigen (alanyl-tRNA synthetase). Am J Med 1990; 88 : 241–51.
14. Targoff IN, Trieu EP, Plotz PH, Miller FW. Antibodies to glycyl-transfer RNA synthetase in patients with myositis and interstitial lung disease. Arthritis Rheum 1992; 35 : 821–30.
15. Hirakata M, Suwa A, Nagai S, Kron MA, Trieu EP, Mimori T, et al. Anti-KS: identification of autoantibodies to asparaginyl-transfer RNA synthetase associated with interstitial lung disease. J Immunol 1999; 162 : 2315–20.
16. Sato S, Kuwana M, Hirakata M. Clinical characteristics of Japanese patinets with anti-OJ (anti-isoleucyl-tRNA synthetase) autoantibodies. Rheumatology 2007; 46 : 842–5.
17. Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Anti-synthetase syndrome: a new autoantibody to phenylalanyl transfer RNA synthetase (anti-Zo) associated with polymyositis and interstitial pneumonia. Rheumatology 2007; 46 : 1005–8.
18. Hengstman GJ, Brouwer R, Egberts WT, Seelig HP, Jongen PJ, van Venrooij WJ, et al. Clinical and serological characteristics of 125 Dutch myositis patiens; myositis specific autoantibodies aid in the differential diagnosis of the idiopathic inflammatory myopathies. J Neurol 2002; 249 : 69–75.
19. Targof IN, Johnson AE, Miller FW. Antibody signal recognition particle in polymyositis. Arthritis Rheum 1990; 33 : 1361–70.
20. Hengstman GJ, Ter Laak HJ, Vree Egberts WT, Lundberg IE, Moutsopoulos HM, Vencovsky J, et al. Anti-SRP autoantibodies, marker of a necrotizing myopathy. Ann Rheum Dis 2006; 65 : 1635–8.
21. Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to be the signal recognition particle: clinical pathological features. J Neurol Neurosurg Psychiatric 2002; 73 : 420–8.
22. Leff RL, Burgess SH, Miller FW, Love LA, Targoff IN, Dalakas MC, et al. Distinct seasonal patterns in the onset of adult idiopathic inflammatory myopathy in patients with anti-Jo-1 and anti-signal recognition particle autoantibodies. Arthritis Rheum 1991; 34 : 1391–6.
23. Wang HB, Zhang Y. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res 2001; 29 : 2517–21.
24. Seelig HP, Moosbrugger I, Ehrfeld H, Fink T, Renz M, Genth E. The major dermatomyositis-specific Mi-2 autoantigen is a presumed helicase involved in trascriptional activation. Arthritis Rheum 1995; 38 : 1389–99.
25. Ge Q, Nilasena DS, O‘Brien CA, Frank MB, Targoff IN. Molecular analysis of a major antigenic region of the 240-kDa protein of Mi-2 autoantigen. J Clin Invest 1995; 96 : 1730–7.
26. Seelig HP, Renz M, Targoff IN, Ge Q, Frank MB. Two forms of the major antigenic protein of the dermatomyositis-specific Mi-2 autoantigen. Arthritis Rheum 1996; 39 : 1769–71.
27. Hengstman GJD, Vree Egberts WTM, Seelig HP, Lundberg IE, Moutsopoulos HM, Doria A, et al. Clinical characteristics of patients with myositis and autoantibodies to different fragments of the Mi-2‚ antigen. Ann Rheum Dis 2006; 65 : 242–5.
28. Ghirardello A, Zampieri S, Iaccarino L, Tarricone E, Bendo R, Gambari PF, Doria A. Anti-Mi-2 antibodies. Autoimmunity. 2005; 38 : 79–83.
29. Selva-O‘Callaghan A, Labrador-Horrillo M, Solans-Laque R, Simeon-Aznar CP, Martínez-Gómez X, Vilardell-Tarrés M. Myositis-specific and myositis-associated antibodies in a series of eighty-eight Mediterranean patients with idiopathic inflammatory myopathy. Arthritis Rheum 2006; 55 : 791–8.
30. Sato S, Hirakata M, Kuwana M, Suwa A, Inada S, Mimori T, et al. Autoantibodies to a 140-kd polypeptide, CADM-140, in Japanese patients with clinically amyopathic dermatomyositis. Arthritis Rheum 2005; 52 : 1571–6.
31. Kaji K, Fujimoto M, Hasegawa M, Kondo M, Saito Y, Komura K, et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: an association with malignancy. Rheumatology (Oxford) 2007; 46 : 25–8.
32. Targoff IN, Mamyrova G, Trieu EP, Perurena O, Koneru B, O‘Hanlon TP, Miller FW, et al. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum 2006; 54 : 3682–9.
33. Gunawardena H, Wedderburn LR, North J, Betteridge Z, Dunphy J, Chinoy H, et al. Clinical associations of autoantibodies to a p155/140 kDa doublet protein in juvenile dermatomyositis. Rheumatology (Oxford) 2008; 47 : 324–8.
34. Chinoy H, Fertig N, Oddis CV, Ollier WE, Cooper RG. The diagnostic utility of myositis autoantibody testing for predicting the risk of cancer-associated myositis. Ann Rheum Dis 2007; 66 : 1345–9.
35. Mimori T, Imura Y, Nakashima R, Yoshifuji H. Autoantibodies in idiopathic inflammatory myopathy: an update on clinical and pathophysiological significance. Curr Opin Rheumatol 2007; 19 : 523–9.
36. Betteridge Z, Gunawardena H, North J, Slinn J, McHugh N. Identification of a novel autoantibody directed against small ubiquitin-like modifier activating enzyme in dermatomyositis. Arthritis Rheum 2007; 56 : 3132–7.
37. Arad-Dann H, Isenberg D, Ovadia E, Shoenfeld Y, Sperling J, Sperling R. Autoantibodies against a nuclear 56 kDa protein: a marker for inflammatory muscle disease. J Autoimmun 1989; 2 : 877–88.
38. Blüthner M, Bautz FA. Cloning and characterization of the cDNA coding for a polymyositis-scleroderma overlap syndrome-related nucleolar 100-kD protein. J Exp Med 1992; 176 : 973–80.
39. Raijmakers R, Renz M, Wiemann C, Egberts WV, Seelig HP, van Venrooij WJ, et al. PM-Scl-75 is the main autoantigen in patients with the polymyositis/scleroderma overlap syndrome. Arthritis Rheum 2004; 50 : 565–9.
40. Mahler M, Raijmakers R, Dähnrich C, Blüthner M, Fritzler MJ. Clinical evaluation of autoantibodies to a novel PM/Scl peptide antigen. Arthritis Res Ther 2005; 7: R704–13.
41. Mimori T, Hardin JA, Steitz JA. Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders. J Biol Chem 1986; 261 : 2274–8.
42. Cavazzana I, Ceribelli A, Quinzanini M, Scarsi M, Airė P, Cattaneo R, et al. Prevalence and clinical associations of anti-Ku antibodies in systemic autoimmune diseases. Lupus 2008; 17 : 727–32.
43. Koenig M, Fritzler MJ, Targoff IN, Troyanov Y, Senécal JL. Heterogeneity of autoantibodies in 100 patients with autoimmune myositis: insights into clinical features and outcomes. Arthritis Res Ther 2007; 9: R78.
44. Vencovský J, Williams DG, Field M, Maini RN. Clinical associations of IgG antibodies to the RNP p-67 polypeptide in patients with systemic lupus erythematosus. Ann Rheum Dis 1992; 51 : 1313–17.
45. Coppo P, Clauvel JP, Bengoufa D, Oksenhendler E, Lacroix C, Lassoued K. Inflammatory myositis associated with anti-U1-small nuclear ribonucleoprotein antibodies: a subset of myositis associated with a favourable outcome. Rheumatology (Oxford) 2002; 41 : 1040–6.
46. Feist E, Dörner T, Kuckelkorn U, Schmidtke G, Micheel B, Hiepe F, et al. Proteasome alpha-type subunit C9 is a primary target of autoantibodies in sera of patients with myositis and systemic lupus erythematosus. J Exp Med 1996; 184 : 1313–8.
47. Wada K, Kamitani T. Autoantigen Ro52 is an E3 ubiquitin ligase. Biochem Biophys Res Commun 2006; 339 : 415–21.
48. Rutjes SA, Vree Egberts WT, Jongen P, Van Den Hoogen F, Pruijn GJ, Van Venrooij WJ. Anti-Ro52 antibodies frequently co-occur with anti-Jo-1 antibodies in sera from patinets with idiopathic inflammatory myopathy. Clin Exp Immunol 1997; 109 : 32–40.
49. Frank MB, McCubbin V, Trieu E, Wu Y, Isenberg DA, Targoff IN. The association of anti-Ro52 autoantibodies with myositis and scleroderma autoantibodies. J Autoimmun. 1999 Mar; 12(2): 137–142.
50. Kajihara M, Kuwana M, Tokuda H, Yamane K, Kubo M, Hirakata M, et al. Myositis and interstitial lung dinase associated with autoantibody to a transfer RNA-related protein Wa. J Rheumatol 2000; 27 : 2707–10.
51. Casciola-Rosen LA, Pluta AF, Plotz PH, Cox AE, Morris S, Wigley FM, et al. The DNA mismatch repair enzyme PMS1 is a myositis-specific autoantigen. Arthritis Rheum 2001; 44 : 389–96.
52. Staněk D, Vencovský J, Kafková J, Raška I. Heterogenous nuclear ribonucleoprotein (hnRNP) C1 a C2 „core“ proteins are targets for an autoantibody found in the serum of a patient with systemic sclerosis and psoriatic arthritis. Arthritis Rheum 1997; 40 : 2172–7.
53. Heegaard NH, Larsen MR, Muncrief T, Wiik A, Roepstorff P. Heterogeneous nuclear ribonucleoproteins C1/C2 identified as autoantigens by biochemic and mass spectrometric methods. Arthritis Rheum 2000, 2 : 407–14.
54. Schilders G, Vree Egberts W, Raijimakers R, Pruijn GJM. C1D is a major antoantibody target in patients with the polymyositis-scleroderma overlap syndrome. Arthritis Rheum 2007; 56 : 2449–54.
55. Oddis CV, Fertig N, Goel A, et al. Clinical and serological characterization of the anti-MJ antibody in childhood myositis. Arthritis Rheum 1997; 40: S139.
56. Mimura Y, Yazawa N, Tamaki Z, Ashida R, Jinnin M, Asano Y, et al. Anti-p53 antibodies in patients with dermatomyositis/polymyositis. Clin Rheumatol 2007; 26 : 1328–31.
57. Targoff IN, Arnett FC, Berman L, O‘Brien C, Reichlin M. Anti-KJ: a new antibody associated with the syndrome of polymyositis and interstitial lung disease. J Clin Invest 1989; 84 : 162–72.
Labels
Dermatology & STDs Paediatric rheumatology RheumatologyArticle was published in
Czech Rheumatology

2008 Issue 4
Most read in this issue
- Specifické a asociované autoprotilátky u polymyozitidy a dermatomyozitidy
- Vrozený srdeční blok plodu způsobený mateřskými autoprotilátkami anti-SSA/Ro a anti-SSB/La
- Hodnocení účinnosti nefarmakologické léčby (pravidelné cvičení), farmakoterapie (glukosamin sulfát, GS Condro Forte®) a kombinace obou postupů u symptomatické gonartrózy. Výsledky otevřené, randomizované, kontrolované studie
- Vztah adiponectinu ke změnám v kůži u pacientů se systémovou sklerodermií