
Vzácné anémie ze skupiny vrozených syndromů selhání kostní dřeně
Authors:
Dagmar Pospíšilová
Authors‘ workplace:
Dětská klinika LF UP a FN Olomouc
Published in:
Vnitř Lék 2018; 64(5): 488-500
Category:
Overview
Přehledný článek přibližuje patofyziologii, molekulárně-genetickou podstatu a klinické projevy anémií ze skupiny vrozeného selhání kostní dřeně s postižením erytroidní linie. Zahrnuje nejnovější poznatky o třech onemocněních, jejichž podstata byla odhalena až v posledních letech díky pokrokům v oblasti molekulární genetiky: Diamondově-Blackfanově anémii, kongenitální dyserytropoetické anémii a Fanconiho anémii. Fanconiho anémie je do přehledu zařazena pro častou přítomnost makrocytózy nebo anémie při manifestaci onemocnění, u většiny pacientů se však postupně vyvíjí selhání všech hematopoetických linií. Cílem přehledu je upozornit současně na méně nápadné příznaky uvedených nemocí, které vedou k jejich přehlédnutí a pozdní diagnostice. V řadě případů je správná diagnóza stanovena až v dospělém věku nebo dokonce až při rozvoji maligního onemocnění. Včasná diagnóza je nezbytná ke stanovení správného léčebného postupu, diagnostice nemoci u členů rodiny, především při vyhledávání dárců kostní dřeně, a ke genetickému poradenství. Cílem práce je rovněž přispět ke včasné diagnostice anémií doprovázených typickými somatickými anomáliemi.
Klíčová slova:
Diamondova-Blackfanova anémie – dyserytropoéza – Fanconiho anémie – kongenitální dyserytropoetická anémie – ribosomopatie – syndromy selhání kostní dřeně – systém oprav DNA – zvýšené riziko vzniku maligního onemocnění
Úvod
Do skupiny vrozených syndromů selhání kostní dřeně (Inherited Bone Marrow Failure Syndromes – IBMFS) jsou řazeny vzácné poruchy krvetvorby charakterizované:
- geneticky podmíněným selháním funkce jedné nebo více hematopoetických linií (erytrocytární, granulocytární nebo trombocytární)
- vrozenými anomáliemi nebo funkčními poruchami postihujícími různé tkáně a orgány: kůži, skelet, srdce, plíce, ledviny, centrální nervový systém, často doprovázené i malým vzrůstem
- zvýšeným rizikem vzniku maligních onemocnění: nejčastěji akutní myeloidní leukemie (AML), myelodysplastického syndromu (MDS) nebo některých typů solidních nádorů
Společným rysem těchto onemocnění je výrazná fenotypová variabilita jak hematologických, tak i somatických změn. Závažnější formy onemocnění se zřetelnými vrozenými anomáliemi, časnou manifestací cytopenií a rozvojem selhání kostní dřeně jsou obvykle diagnostikovány již v časném dětství v kojeneckém, batolecím nebo předškolním věku. Lehčí formy s nenápadnými anomáliemi a pozdní manifestací cytopenií mohou na druhé straně dlouho unikat pozornosti a jsou potom diagnostikovány až v dospělosti, nebo dokonce až při vypuknutí maligního onemocnění. Tuto skupinu onemocnění dělíme obvykle podle nejvýrazněji postižené linie krvetvorby a rozsahu jejich postižení na izolované cytopenie a pancytopenie.
U izolovaných cytopenií (Diamondova-Blackfanova anémie, kongenitální dyserytropoetická anémie, těžká vrozená neutropenie, trombocytopenie s chyběním radia) nedochází obvykle k rozvoji pancytopenie, u ostatních onemocnění se postupně rozvíjí pancytopenie s aplazií kostní dřeně (Fanconiho anémie, dyskeratosis congenita, Shwachmanův-Diamondův syndrom, kongenitální amegakaryocytární trombocytopenie).
Do skupiny vrozených syndromů setkání kostní dřeně jsou dnes řazena následující onemocnění:
- Fanconiho anémie (FA)
- dyskeratosis congenita (DC)
- syndrom hypoplastických chrupavek a vlasů (Cartilage-Hair Hypoplasia – CHH)
- Diamondova-Blackfanova anémie (DBA)
- Shwachmanův-Diamondův syndrom (SDS)
- těžká vrozená neutropenie (Severe Congenital Neutropenia – SCN)
- retikulární dysgeneze (RD)
- amegakaryocytární trombocytopenie (Congenital Amegakaryocytic Thrombocytopenia – CAMT)
- trombocytopenie s chyběním radia (thrombocytopenia with absent radius – TAR)
- Pearsonův syndrom (PS)
- kongenitální dyserytropoetická anémie (Congenital Dyserythropoietic Anemia – CDA) – tato byla zařazena do skupiny IBMFS teprve v posledních letech
Epidemiologie
Všechna tato onemocnění jsou vzácná, incidence některých z nich není přesně známa. V řadě případů je regionálně odlišná a závislá na lokální frekvenci nosičství mutací příslušných genů. V některých uzavřených populačních skupinách je vyšší frekvence výskytu v důsledku tzv. efektu zakladatele (founder effect), což znamená ztrátu genetické variability, která nastává při vytvoření uzavřené nové populace s malým počtem jedinců. Typ dědičnosti je u jednotlivých chorob variabilní a závisí na charakteru vyvolávajícího genetického defektu. Při významném zlepšení diagnostických možností se v posledním desetiletí zvyšuje i počet diagnostikovaných případů. Předpokládá se tedy, že tato onemocnění jsou nedostatečně diagnostikována, a to především jejich mírnější formy. Včasné stanovení diagnózy a určení genetického defektu je přitom velmi důležité pro stanovení adekvátního plánu dlouhodobého sledování pacienta, léčebného postupu a prognózy onemocnění. Má velký význam i pro genetické poradenství a vyhledávání dalších postižených členů rodiny [1].
Vrozené syndromy kostní dřeně s postižením erytroidní linie
Diamondova-Blackfanova anémie
Charakteristika
Diamondova-Blackfanova anémie (DBA) je vzácná vrozená hypoplazie erytropoézy, charakterizovaná normochromní makrocytární anémií, těžkou retikulocytopenií, normocelulární kostní dření se selektivním nedostatkem erytroidních prekurzorových buněk, normálním počtem leukocytů a normálním nebo lehce vyšším počtem trombocytů [2]. U 40–50 % pacientů jsou přítomny přídatné anomálie (tab. 1), asi u 30 % pacientů růstová retardace. Riziko rozvoje hematologických maligních onemocnění (MDS a AML) a solidních nádorů je 5–6krát vyšší než u zdravé populace [3].

DBA patří mezi nejčastěji se vyskytující onemocnění ve skupině syndromů selhání kostní dřeně. Poprvé byla popsána jako klinická jednotka v roce 1938 americkými pediatry Louisem K. Diamondem, považovaným za zakladatele pediatrické hematologie, a Kennethem Blackfanem.
Diagnóza je v 90 % stanovena v novorozeneckém nebo kojeneckém věku. Méně často bývá onemocnění zjištěno později, v posledních letech není výjimkou stanovení diagnózy u pacientů s mírnější formou onemocnění až v dospělosti [2]. Výjimečně může být nemoc odhalena až při diagnostice maligního onemocnění v dospělém věku [2], což potvrzuje i vlastní zkušenost autorky. Rozvoj selhání kostní dřeně je vzácný.
Epidemiologie a dědičnost
Incidence je udávána v rozmezí 5–8 případů na 1 milion živě narozených. DBA se vyskytuje u mužského a ženského pohlaví v poměru 1,1 : 1. Většinu pacientů tvoří příslušníci indoevropské populace, nemoc však byla popsána i u africké negroidní populace, Japonců a Arabů.
Ve 20–40 % případů se jedná o hereditární onemocnění s autosomálně dominantním typem dědičnosti. U nově popsaných mutací v extraribosomálních genech kódujících GATA1 transkripční faktor a TSR2 protein je dědičnost vázaná na chromosom X. Ve zbývajících případech se jedná o sporadicky se vyskytující onemocnění, v jejichž etiologii se uplatňují mutace vznikající de novo.
Klinické a laboratorní nálezy
U většiny pacientů je diagnóza stanovena v 1. roce života, u části pacientů je anémie vyžadující podání transfuze diagnostikována již v novorozeneckém věku. Prvními klinickými příznaky DBA jsou bledost, dušnost, u kojenců je často popisováno neprospívání. Vzácně byl popsán i fetální hydrops. U 30–50 % pacientů s DBA jsou přítomny vrozené anomálie, postihující převážně oblast lbi a obličeje (kraniofaciální dysmorfie, mikrocefalie, rozštěp patra, hypertelorizmus a gotické patro). K dalším anomáliím patří vývojové vady palce, horní končetiny (tříčlánkový palec, duplikace článku palce nebo hypoplazie palcového valu (thenar), srdce, ledvin, urogenitálního traktu a kostí (tab. 1, obr. 1, obr. 2).
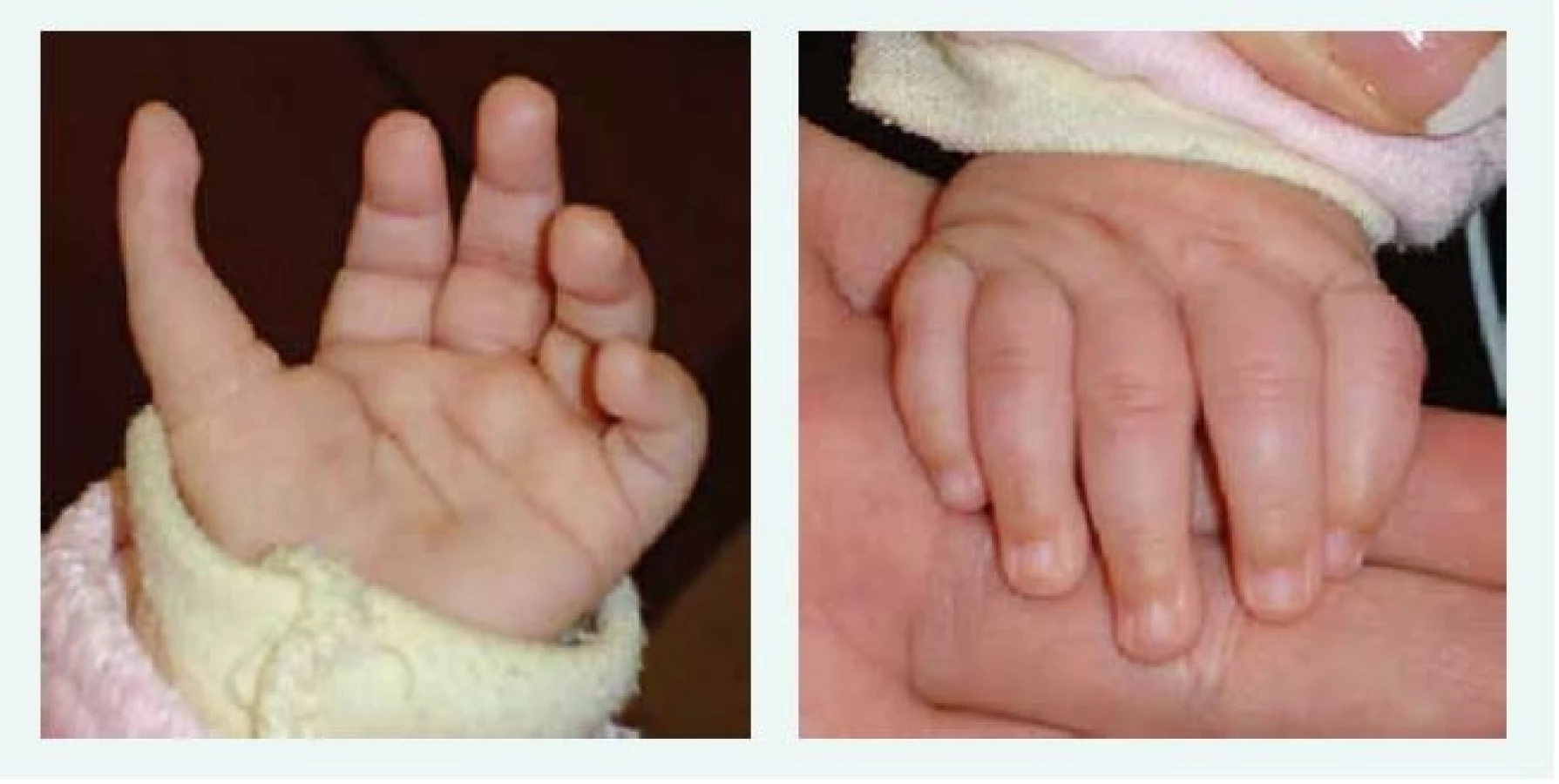

U 30–40 % pacientů je popisován malý vzrůst. Na růstové retardaci u starších pacientů se může podílet dlouhodobá léčba kortikosteroidy i přetížení organizmu železem.
Při laboratorním vyšetření je dominujícím nálezem makrocytární anémie s retikulocytopenií. Může být zvýšená hladina fetálního hemoglobinu (HbF) jako známka stresové erytropoézy. Důležitým znakem je zvýšení hladiny erytrocytární adenozindeaminázy (e-ADA), které může být jedním z prvních diagnostických vodítek. I přes specificitu tohoto nálezu pro DBA nebyla dosud příčina zvýšení e-ADA vysvětlena. V kostní dřeni je v typických případech výrazně snížen počet erytroidních prekurzorových buněk, často na 1–5 %.
Podle analýzy údajů z registru DBA v USA (DBAR) je riziko rozvoje solidních nádorů nebo leukemie u DBA 5,4krát vyšší než v populaci [3]. Zvýšená incidence byla popsána specificky pro některé typy maligních onemocnění: MDS, AML, karcinom plic, tlustého střeva, bazaliom, osteosarkom, nádory prsu a gynekologické karcinomy u žen. Dle DBAR je incidence solidních nádorů vyšší než výskyt leukemií. Kumulativní incidence solidních nádorů a AML u DBA byla průměrně 20 % ve 4. decéniu ve srovnání se 30 % u FA a DC kdykoliv ve stejné věkové kategorii; riziko solidních nádorů u netransplantovaných pacientů začíná narůstat po 30. roce věku, tedy později než u FA a DC. Malý počet popsaných případů a značná diverzita typů maligních onemocnění však zatím nedovoluje definitivní závěry.
Molekulární podstata onemocnění
Po desetiletích snahy o nalezení příčiny DBA byla teprve v roce 1999 poprvé odhalena její genetická podstata: překvapivý nález heterozygotní mutace genu kódujícího ribosomální protein (RP) RPS19, součásti malé ribosomální podjednotky u švédského pacienta. Mutace genu kódujícího RPS19 byly postupně prokázány u 25 % pacientů s DBA. RPS19 je komponentou malé 40S ribosomální podjednotky a je lokalizován v nukleolu, hlavním buněčném místě transkripce při biogenezi ribosomů. Role některých RP není dosud u vyšších eukaryont přesně známa. Delece jedné alely RP vede k poruše růstu a snížené tvorbě 40S ribosomální podjednotky, tedy k tzv. „ribosomálnímu stresu“. Buňka reaguje aktivací p53 proteinu a indukcí apoptózy. Dalším důsledkem je snížení translace [5], a tím i proteosyntézy, což může ovlivnit procesy s velkým nárokem na přísun proteinů – tedy s rychlým obratem produkce buněk, jako je hematopoéza, obnova kožních buněk a buněk GIT.
V dalších letech byly postupně nalezeny mutace genů kódujících dalších 20 RP: RPS7, RPS10, RPS15A, RPS17, RPS19, RPS24, RPS26, RPS27, RPS28, RPS29; RPL5, RPL9, RPL11, RPL15, RPL18, RPL26, RPL27, RPL31, RPL35, and RPL35A [5]. Ojediněle byly nalezeny mutace dvou dalších genů, které nekódují RP: GATA1, důležitého transkripčního faktoru pro erytropoézu a TSR2, který kóduje protein ovlivňující apoptózu a maturaci ribosomů. DBA tedy vzniká v důsledku haploinsuficience ribosomálních proteinů. Aktivace p53 proteinu i snížení translace bylo prokázáno na buněčných [5] i zvířecích modelech DBA. Jsou dále intenzivně studovány extraribosomální funkce RP a jejich možná úloha při vzniku somatických anomálií.
Odhalení poruchy biogeneze ribosomů jako příčiny hematologického onemocnění patří mezi nejzajímavější objevy moderní hematologie a otevřelo mnoho otázek týkajících se souvislosti poruch ribosomální biogeneze, somatických anomálií a zvýšené incidence vzniku maligního onemocnění. Diamondova-Blackfanova anémie tak dala vznik nové skupině onemocnění nazvaných ribosomopatie.
Diagnostika
Pro diagnózu DBA svědčí makrocytární anémie s retikulocytopenií při normálním počtu trombocytů a leukocytů. V době stanovování diagnózy se výjimečně může objevit trombocytóza a/nebo neutropenie. Pro diagnostiku je stěžejní vyšetření kostní dřeně, v níž je klasickým nálezem nízký počet erytroidních prekurzorových buněk v jinak normocelulární kostní dřeni. U pacientů, kteří nejsou závislí na transfuzní terapii ovlivňující výsledky vyšetření, je přínosné vyšetření erytrocytární adenozindeaminázy (eADA), jejíž hodnota je zvýšena. Normální hladina e-ADA nevylučuje onemocnění. Jedinou metodou, která jednoznačně potvrzuje diagnózu, je nález mutací genů pro ribosomální proteiny, případně genu kódujícího TSR2 protein a GATA1 transkripční faktor.
Při diagnostice je vždy nutné vyloučit některá jiná onemocnění, o kterých lze diferenciálně diagnosticky uvažovat. Především se jedná o: Fanconiho anémii, deficit G-6PD, erytroblastopenii vzniklou po infekci parvovirem B19, tranzientní erytroblastopenii dětského věku (Transient Erythroblastopenia of Childhood – TEC) a další získané erytroblastopenie, u pacientů s DBA v remisi s normálním počtem erytroidních prekurzorů, vzácně i kongenitální dyserytropoetickou anémii vzhledem k dysplastickým změnám erytropoézy.
Léčba
Léčebně je v 1. roce života obvykle nezbytné podávání transfuzí erytrocytů. Po 1. roce je indikováno podání kortikoidů, na které až 60–80 % pacientů odpoví akcelerací erytropoézy. Kortikoidy se podávají nejprve po dobu 4 týdnů v dávce 2 mg/kg/den, při dobré odpovědi je potom dávka prednisonu postupně pomalu snižována s cílem dosažení obdenní dávky nepřesahující 0,5 mg/kg. Dlouhodobé podávání denních dávek vyšších než 0,5 mg/kg se nedoporučuje vzhledem k riziku významných vedlejších účinků v dětském věku: rozvoj osteoporózy, přírůstek na váze, cushingoidní vzhled, rozvoj hypertenze a diabetu, růstová retardace, patologické fraktury, vznik žaludečních vředů, katarakty nebo glaukomu a zvýšenou náchylnost k infekcím.
U pacientů, kteří neodpovídají příznivě na léčbu kortikoidy, je nutno podávat dlouhodobě transfuze erytrocytární masy. Cílem transfuzního programu je udržení koncentrace hemoglobinu mezi 80–100 g/l, což je hladina adekvátní pro udržení normálního růstu a vývoje dítěte [2].
Z celkového počtu DBA pacientů je přibližně 40 % pacientů steroid-dependentních, asi 40 % je závislých na transfuzích a 20 % dosáhne remise.
U pacientů, kterým jsou podávány pravidelně transfuze erytrocytární masy, je nezbytné podávání chelátorů železa (desferioxamin, deferasirox) k prevenci orgánového postižení při přetížení železem. Léčbu chelátory je vhodné zahájit již po podání prvních 12 transfuzí, po nichž dosáhne sérový feritin obvykle hladin 1 000–1 500 µg/l, nebo když koncentrace železa v jaterní tkáni dosáhne 6–7 mg/g sušiny jaterní tkáně. Podávání deferasiroxu je vhodné až od 2 let věku. Deferipron běžně používaný k léčbě přetížení železem u pacientů s talasemií major není pro dlouhodobou léčbu u DBA doporučován pro zvýšené riziko vzniku agranulocytózy [6]. Je nutno sledovat pečlivě růst pacienta a monitorovat možné nežádoucí účinky přetížení železem i chelatační léčby. Nedílnou součástí týmu pečujícího o pacienty s DBA je proto i endokrinolog, jehož úkolem je včas podchytit a léčit poruchy růstu, kostní denzity, fertility, onemocnění štítné žlázy a hypofýzy vznikající v důsledku léčby kortikoidy a/nebo přetížení železem. U části pacientů (asi 20–30 %) může nastat remise onemocnění (spontánně nebo při opakované léčbě kortikoidy), která nastupuje obvykle do 8.–10. roku věku, velmi vzácně i později.
Zkoumá se i efekt nových látek s pozitivním efektem na erytropoézu s výhledem léčebného využití: aminokyseliny leucinu [7,8] a sotaterceptu [9].
Pokud lze nalézt v rodině pacienta příbuzenského dárce, případně shodného dárce v rámci registrů, je u pacientů trvale závislých na podávání transfuzí indikována transplantace kmenových buněk (Hematopoietic Stem Cell Transplantation – HSCT), která je zatím jedinou kurativní léčbou pro DBA. Pacienti trvale závislí na transfuzích nebo ti, u kterých se vyvinuly další cytopenie, jsou indikováni k HSCT. Otázka vhodných režimů a optimálního věku transplantace je předmětem diskusí, zatím se její provedení doporučuje do 10 let věku. HSCT od nepříbuzenských dárců má horší výsledky, i když v posledních letech došlo k jejich významnému zlepšení.
V průběhu těhotenství žen s Diamondovou-Blackfanovou anémií byly popsány četné komplikace preeklampsie, intrauterinní růstová retardace a úmrtí plodu, zhoršení anémie, předčasné porody [10].
Kongenitální dyserytropoetické anémie
Charakteristika
Kongenitální dyserytropoetické anémie (Congenital Dyserythropoietic Anemias – CDA) zahrnují skupinu vzácných vrozených poruch erytropoézy charakterizovaných chronickou hyporegenerativní anémií s neefektivní erytropoézou, relativní retikulocytopenií, hemolytickou složkou s mírnou hyperbilirubinemií a splenomegalií, typickými morfologickými změnami erytroblastů v kostní dřeni a v některých případech i přetížením železem. Většinou se jedná o poruchy vyzrávání erytroidních buněk v pozdních stadiích erytropoézy. V dnešní době je CDA řazena většinou hematologů do skupiny syndromů selhání kostní dřeně vzhledem k poruše diferenciace a proliferace buněk erytroidní linie [11].
Termín dyserytropoéza označuje kvalitativní defekt erytroidních prekurzorů nebo erytrocytů vedoucí k abnormální (neefektivní) erytropoéze s postižením diferenciace a proliferace erytroidních buněk, které v kostní dřeni nevyzrávají nebo částečně zanikají před vyplavením do periferní krve. V některých případech je doprovázena i zkráceným přežíváním erytrocytů. Dyserytropoéza provází řadu jak vrozených (talasemie a jiné hemoglobinopatie, hereditární sférocytóza, sideroblastická anémie, DBA, FA), tak získaných hematologických onemocnění (deficit železa, vitaminů, MDS), což může být zdrojem diferenciálně diagnostických rozpaků a obtížné diagnostiky.
Epidemiologie a dědičnost
CDA patří mezi vzácné anémie. Prevalence a geografická distribuce se liší mezi jednotlivými oblastmi. Nejnižší výskyt je popisován v severských zemích (0,04 případů na 1 milion obyvatel), nejvyšší výskyt v Itálii (2,4 případu na 1 milion obyvatel) [12]. Některé mutace se vyskytují s vysokou frekvencí v určitých populacích (u beduínské populace R1042W v genu pro codanin – CDAN1, v populaci zemí kolem Středozemního moře – Maroko, Izrael, Itálie – R14W v genu SEC23B). Výskyt jednotlivých alel však svědčí o vlivu mnohočetného efektu zakladatele (founder effect) v jednotlivých geografických oblastech.
Dědičnost jednotlivých typů CDA je většinou autosomálně recesivní, v některých případech dominantní.
Klinické a laboratorní nálezy
Klinicky se CDA projevují celoživotní středně těžkou až těžkou makrocytární nebo normocytární anémií s časně vznikajícím ikterem, splenomegalií a hepatomegalií. Často se u pacientů tvoří žlučové kameny. V některých případech může být prvním projevem onemocnění fetální hydrops nebo intrauterinní růstová retardace. U 4–14 % pacientů jsou přítomny vrozené anomálie, především v oblasti skeletu končetin. V důsledku opakovaných transfuzí a zvýšené resorpce železa se u polytransfundovaných pacientů, ale i části pacientů bez nutnosti transfuzí se rozvíjí přetížení železem se sekundární hemochromatózou. Jednou z příčin je i nízká hladina hepcidinu.
Při laboratorním vyšetření nacházíme v krevním obrazu normocytární nebo makrocytární anémii spojenou s anizocytózou, poikilocytózou, nálezem bazofilního tečkování erytrocytů, Howellových-Jollyových tělísek a relativní retikulocytopenií. V kostní dřeni je typická hyperplazie erytropoézy s výraznými dysplastickými změnami: dvoujaderné a mnohojaderné erytroblasty, internukleární můstky, vakuolizace cytoplazmy, nepravidelná kontura jader (obr. 3).

Typické změny jsou popisovány při vyšetření elektronovou mikroskopií (obr. 4).

Podezření na CDA tedy vychází z klinického obrazu a hematologických nálezů v periferní krvi. Vždy je nutno provést vyšetření kostní dřeně se zhodnocením morfologie erytroblastů. Významnou roli v diagnostice hraje i elektronová mikroskopie. Diagnóza je definitivně potvrzena nálezem patogenních variant pomocí molekulárně-genetického vyšetření, které jsou typické pro jednotlivé subtypy onemocnění (tab. 2).

Diagnóza
Pracovní klasifikace navržená poprvé Heimpelem a Wendtem (CDA I, II, a III) je stále v klinické praxi používána [13]. V některých případech anémie splňuje obecnou definici CDA, nelze ji však zařadit do žádného ze základních 3 subtypů, proto byly tyto označeny jako tzv. CDA varianty. Molekulárně genetický podklad obou nejčastějších typů: CDA Ia a II je znám již delší dobu, kauzální genetické změny u typu III, IV a u ostatních CDA variant však byly popsány teprve v posledních letech [6,7,9], což výrazně zpřesnilo diagnostiku této skupiny anémií.
Molekulární podstata onemocnění
Vzhledem k významným pokrokům v oblasti molekulární biologie, které přináší především sekvenování nové generace, byly dnes již u jednotlivých typů CDA identifikovány kauzální molekulárně-genetické změny, což významně přispělo k upřesnění klasifikace u této skupiny nemocí, a především k postupnému porozumění jejich patogenezi. Podle genetických změn je dnes rozlišováno 5 základních podtypů CDA (Ia, Ib, II, III a IV), tab. 3.
![Vrozené vývojové anomálie u pacientů s Fanconiho anémií. Upraveno podle [31]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/e133b864cbf217f4d2cdf7efcdf458a8.jpeg)
Některé další varianty dyserytropoetické anémie jsou součástí komplexních onemocnění či syndromů a jsou některými autory označované jako typ V a VI. S objevem kauzálních genetických variant se otevřela nová éra genetického a klinického výzkumu s cílem detailního odhalení funkce proteinů, v jejichž kódujících genech jsou popisovány mutace. Stanovení molekulární podstaty dnes umožňuje diagnostiku nosičství genu i prenatální diagnostiku, může být vodítkem při hledání nových alternativ léčby. Molekulárně-genetické charakteristiky jsou uvedeny u jednotlivých podtypů CDA.
Kongenitální dyserytropoetická anémie typu I
Kongenitální dyserytropoetická anémie typu I (CDA I) je autosomálně recesivně dědičná anémie charakterizovaná makrocytární anémií MCV (100–120 fL) a u některých pacientů i anomáliemi distálních částí končetin. Při vyšetření kostní dřeně je přítomna erytroidní hyperplazie s přítomností méně než 10 % dvojjaderných erytroblastů. Typickou změnou jsou internukleární můstky přítomné v 5–8 % erytroblastů. Elektronová mikroskopie ukazuje houbovitý vzhled heterochromatin označovaný jako „vzhled švýcarského sýra“ („Swiss cheese appearance“).
Většina pacientů s CDA I má makrocytární anémii s variabilní hladinou hemoglobinu, výjimečně může být anémie v dětském věku normocytární. Relativní retikulocytopenie je doprovázena zvýšeným rozpadem erytrocytů se zvýšenou hladinou nepřímého bilirubinu, vysokou hladinou laktátdehydrogenázy (LDH), snížením hladiny haptoglobinu a hyperbilirubinemií. Všichni pacienti mají od adolescentního věku splenomegalii, 20 % dětí má vrozené anomálie, nejčastěji syndaktylie na rukou nebo nohou, chybění nehtů nebo nadpočetné palce, deformity hrudníku a malý vzrůst. Byly popsány i deformity lebky v důsledku výrazné expanze kostní dřeně. Výjimečně byl popsán fetální hydrops a anémie vyžadující intrauterinní transfuzi. Asi 1 ze 3 plodů je menší než odpovídá jeho gestačnímu věku. Po porodu může být přítomna hepatomegalie a ikterus, v některých případech je nutná i transfuze erytrocytů.
Příčinou vzniku jsou mutace CDAN1 genu kódujícího codanin, které byly popsány v roce 2003 [14]. Dle dosavadních poznatků hraje tento protein důležitou roli v organizaci jaderného heterochromatinu v průběhu replikace DNA. Codanin 1 je rovněž součástí cytosolického Asf1-H3-H4-importin-4 komplexu, který má důležitou funkci při sestavování a degradaci nukleosomu. Pravděpodobně hraje roli i v časné fázi vývoje embrya.
Nález kauzálních mutací dalšího genu, C15ORF41, u pacientů s CDA I vedl k rozdělení CDA I na 2 podtypy: CDA 1a a 1b (tab. 2). Tento gen kóduje restrikční endonukleázu s dosud neznámou funkcí. Bylo však prokázáno, že C15ORF41 kooperuje s Asf1b, což podporu hypotézu, že primárním defektem u CDA 1b je rovněž patologie DNA replikace a sestavování chromatinu [15].
Kongenitální dyserytropoetická anémie typu II
CDA II je nejčastější variantou CDA. V kostní dřeni je více než 10 % binukleárních erytroblastů, elektronová mikroskopie ukazuje vezikuly s obsahem proteinů endoplazmatického retikula lokalizované pod plazmatickou membránou. U CDA II je pozitivní Hamův test (lýza erytrocytů s acidifikovaným alogenním sérem), který byl dříve používán k diagnostickým účelům. Vzhledem k potřebě několika kontrolních vzorků se dnes již běžně nepoužívá.
Analýza proteinů erytrocytární membrány SDS PAGE elektroforézou na polyakrylamidovém gelu, která identifikuje abnormality glykosylace rychle migrujícího pruhu 3 (the anion exchanger 1) a pruhu 4,5, je vysoce senzitivní a specifická. K potvrzení diagnózy je používána molekulárně-genetická analýza. V roce 2012 byla poprvé popsána molekulární příčina onemocnění: mutace genu SEC23B kódujícího protein COPII, jenž je součástí proteinového komplexu podílejícího se na tvorbě a transportu vezikul v buňce [16]. Teprve po několika letech však byl objasněn patogenetický mechanizmus vzniku patologických změn erytroblastů u CDA II.
Fenotyp multinuklearity vzniká při aberantní glykosylaci specifických proteinů nezbytných pro dělení buněk, což vede k defektům v tomto procesu. Specificita efektu na erytropoézu u takto ubikvitního genu je pravděpodobně způsobena tkáňově specifickou expresí SEC23B během erytroidní diferenciace.
Kongenitální dyserytropoetická anémie typu III
CDA III je nejvzácnějším typem CDA. Poprvé byla popsána v rodině z USA, většina pacientů jsou členové jedné velké rodiny ze Švédska. Dědičnost je v obou rodinách dominantní. Bylo však popsáno i několik případů s autosomálně recesivní dědičností. Splenomegalie většinou není přítomna. Nález v kostní dřeni je charakterizován erytroidní hyperplazií a velkými typickými multinukleárními erytroblasty. U mnoha členů švédské rodiny byla diagnostikována monoklonální gamapatie a u některých došlo k rozvoji mnohočetného myelomu.
Příčinou onemocnění je mutace genu KIF23 kódujícího MKLP1 protein, což je mitotický protein důležitý pro cytokinezi [17,18]. Porucha funkce tohoto proteinu vede ke vzniku multinuklearity erytroblastů.
Kongenitální dyserytropoetická anémie typu IV
CDA IV je velmi vzácným typem této skupiny. Kauzální jsou mutace genu KLF1 kódujícího transkripční faktor [19,20]. V roce 2010 byla u pacienta s CDA poprvé popsána mutace genu KLF1 lokalizovaná v exonu 3 (jedná se o mutaci měnící smysl: c.973 G>A, p.E325K). Dosud byly popsány pouze 4 případy onemocnění, 5. dosud nepublikovaný případ stejné mutace byl popsán v České republice.
Krüppel-like factor 1 (KLF1) je důležitý transkripční faktor v procesu erytropoézy. Účastní se regulace vyzrávání multipotentních kmenových buněk do erytroidní linie, hemoglobinového přepínání z fetálního na adultní hemoglobin a pozdní diferenciaci erytroidních buněk. Je důležitý pro udržování buněčné stěny v klidovém období života erytrocytů. Poslední studie ukázaly, že mutace v některých lokusech KLF1 genu se vyskytují poměrně často a jsou většinou asymptomatické, vedou však ke zvýšení hladin fetálního hemoglobinu (α2γ2) a adultního HbA2 (α2δ2).
Kongenitální dyserytropoetické anémie variantní
Do dnešního dne bylo popsáno několik dalších CDA variant. Tato onemocnění mají buďto izolovaný CDA fenotyp nebo se jedná o definované syndromy, u kterých je přítomen různý rozsah dyserytropoézy. Do této skupiny jsou řazeny následující jednotky:
- Makrocytární dyserytropoetická anémie s dominantní dědičností vázanou na chromosom X: charakterizovaná zvýšenou hladinou feritinu (800 ng/ml) – byla popsána u několika osob ženského pohlaví v jedné rodině – v kostní dřeni je výrazná dyserytropoéza, bez známek kumulace železa nebo sideroblastů; příčinou je překvapivě mutace genu pro ALAS2 (Y365C), která se podílí na syntéze hemu, recesívní mutace ALAS2 byly doposud asociovány se sideroblastickou anémií s dědičností vázanou na X chromosom (tzv. XLSA)
- Trombocytopenie s, nebo bez dyserytropoetické anémie s recesivní dědičností vázanou na chromosom X (XLTDA), charakterizovaná anémií různé závažnosti od hydrops fetalis a závislosti na transfuzích k projevům dyserytropoézy bez anémie, makrotrombocytopenií s hypogranulovanými trombocyty a tendencí ke krvácení, až absenci demarkace trombocytární membrány – příčinou jsou mutace genu kódujícího transkripční faktor GATA1 lokalizovaného na chromosomu X. GATA1 transkripční faktor hraje důležitou roli ve vývoji a udržování buněk erytroidní a megakaryocytární linie, o rozdílném efektu různých typů mutací svědčí různé fenotypy onemocnění, které dosud byly popsány podobně jako např. v případě KLF1 transkripčního faktoru – různé typy popsaných mutací GATA1 se přitom vyznačují různým fenotypem onemocnění, což je způsobeno rozdílným efektem jednotlivých mutací na funkci GATA1 transkripčního faktoru
- Kongenitální dyserytropoetická anémie byla popsána i jako součást klinického obrazu Majeed syndromu, vzácného autosomálně recesivního onemocnění charakterizovaného časným rozvojem chronické rekurentní multifokální osteomyelitidy, dermatitidami a hypochromní mikrocytární anémií s dyserytropoézou, erytroidní hyperplazií s binukleárními a trinukleárními erytroblasty – onemocnění je způsobené mutacemi genu LPIN2, kódujícího lipin 2, fosfatázu důležitou pro metabolizmus lipidů
- Dyserytropoetické anémie spojené s exokrinní pankreatickou dysfunkcí a hyperostózami lebky, způsobené mutacemi genu COX4I2 kódujícího podjednotku cytochrom C oxidázy (COX) podílející se na dýchacím řetězci v mitochondriích
- Těžká dyserytropoetická anémie spojená s recesívně dědičným deficitem mevalonát kinázy (MVK) v důsledku mutace genu pro MVK
Diferenciální diagnóza
Dyserytropoéza je často přítomna u anémií při deficitu železa, folátu, vitaminu B6 a E v podmínkách s relativní hypoxií jako je neonatální období, vrozené srdeční vady, chronická onemocnění bronchů, kdy se může objevit sekundární dyserytropoéza. Dyserytropoetické změny jsou přítomny i v kostní dřeni pacientů s vrozenými syndromy selhání kostní dřeně, především u DBA a Fanconiho anémie. Jsou jedním z diagnostických znaků u myelodysplastického syndromu. Všechna tato onemocnění musí být před stanovením diagnózy vyloučena.
Léčba
Při těžké anémii je nutné pravidelné podávání transfuzí, intramuskulární nebo subkutánní podání interferonu (IFN), a to INFα2a nebo INFα2b podávaného 2–4krát týdně. To vede u většiny pacientů s CDA I. typu ke zvýšení hladiny Hb a snížení přetížení železem [21,22]. U několika pacientů s těžkou formou CDA byla provedena úspěšná alogenní transplantace kmenových buněk, která by však měla být zvažována pouze u dětí závislých na transfuzích, které nereagovaly na podávání interferonu [23,24].
U pacientů v pravidelném transfuzním programu je nutná prevence sekundárních komplikací, především důsledné monitorování přetížení železem (každé 3 měsíce) včetně ročního vyšetření myokardiálního T2* MRI a jaterního R2* MRI od 10 let věku. Nezbytná je důsledná medikamentózní léčba přetížení železem za použití dostupných chelátorů železa (desferioxamin, deferipron, deferasirox). Pravidelné flebotomie jsou vzhledem k anémii nevhodné.
Závěr
Vzhledem k obrovskému pokroku v odhalování genetické příčiny CDA byly analyzovány kauzální mutace vedoucí ke vzniku CDA a vysvětlena jejich patogenetická role při vzniku tohoto onemocnění. To s sebou přináší i možnosti genetického poradenství, a to jak vyhledávání patologických mutací u členů rodiny, tak možnost prenatální diagnostiky a částečně i některé možnosti léčby jako je použití interferonu α u CDA.
V České republice bylo diagnostikováno dosud okolo 15 pacientů s CDA.
Fanconiho anémie
Charakteristika
Fanconiho anémie (FA) je klinicky i geneticky výrazně heterogenní onemocnění charakterizované postupně progredujícím selháním kostní dřeně, přítomností různých kombinací somatických anomálií a zvýšeným výskytem maligních onemocnění: MDS, AML a solidních nádorů rezistentních na léčbu. Prvním příznakem bývá trombocytopenie nebo anémie.
Poprvé byla popsána v roce 1927 švýcarským pediatrem Guidem Fanconim [25], molekulární podstata onemocnění byla postupně odhalena v 90. letech minulého století.
Epidemiologie a dědičnost
Incidence onemocnění je 1–5 na 1 milion živě narozených, přičemž jsou popsány výrazné regionální rozdíly. Frekvence nosičů genu je v populaci USA odhadována na 1 : 181, vyšší je popisována např. v populaci aškenázských (středo - a východoevropských) Židů (1 : 93) [26] a u černošské populace v malarických oblastech subsaharské Afriky, v nichž je incidence FA udávána asi u 1 na 40 000 živě narozených, což je významně vyšší než se původně předpokládalo [27]. Kauzální delece FANCG je zde zřejmě „founder mutation“ u populace kmene Bantu.
Onemocnění je autosomálně recesivně dědičné kromě jednoho z genetických defektů (FANCB) u kterého je dědičnost gonosomální vázaná na chromosom X.
Klinické a laboratorní nálezy
Na onemocnění může již v útlém věku upozornit přítomnost pestré palety vrozených anomálií. Jsou popisovány u 80 % pacientů, patří k nim: malý vzrůst spojený často s intrauterinní růstovou retardací, hyperpigmentace kůže, malformace palce, skeletu, ledvin, srdce, poruchy sluchu, a hypogonadizmus (tab. 3).
Selhání KD se klinicky manifestuje obvykle kolem 7. roku věku trombocytopenií, anémií, postupně se rozvíjí pancytopenie. Asi ve 4 % případů se aplazie kostní dřeně rozvíjí již během kojeneckého věku. Vzácně se naopak může cytopenie objevit až v dospělosti, někdy až současně s rozvojem maligního onemocnění. U 50 % pacientů, kteří se prezentují trombocytopenií, dochází k rozvoji pancytopenie v průběhu následujících 4 let, kumulativní incidence rozvoje selhání kostní dřeně je 50–90 % ve věku 40 let.
Některé literární zdroje uvádějí, že u 25–40 % jedinců s FA jsou vrozené anomálie nenápadné, nebo dokonce nejsou přítomny vůbec. Totéž platí pro známky selhání kostní dřeně, které mohou být pouze diskrétní – mírná anémie nebo trombocytopenie, a mohou tak dlouho unikat pozornosti [28,29].
U pacientů s FA je 50krát vyšší riziko vzniku maligních onemocnění ve srovnání se zdravou populací: kumulativní incidence MDS, AML a solidních nádorů ve věku 40 let je 30,10 a 30 % [7]. Nejčastějšími solidními tumory jsou spinocelulární karcinomy hlavy, krku, jazyka, jícnu a vulvy.
Molekulární podstata onemocnění
Charakteristickou vlastností buněk pacientů s FA je chromosomální nestabilita, kterou je možno in vitro dále akcentovat přidáním látek navozujících klastogenní stres (mitomycin C, diepoxybutan). Působení těchto látek indukuje vznik chromosomálních zlomů a fúzí s charakteristickými radiálními formami. Popisuje se i zvýšená tendence k apoptóze a zvýšená náchylnost k toxickému působení volných kyslíkových radikálů. Pestrému a variabilnímu klinickému obrazu odpovídá i výrazná heterogenita vyvolávajících genetických defektů, z nichž většina byla postupně analyzována až v posledních letech. Jednotlivé geny kódují proteiny, které jsou složkami systému reparace DNA. Geny, jejichž mutace byly popsány u pacientů s FA, se nazývají FANC geny – tzv. FANC komplementační skupiny Jejich genové produkty regulují 3 důležité procesy v průběhu oprav meziřetězcových křížových vazeb DNA: nukleolytické incize, translézní opravu DNA a homologní rekombinaci. Buňky pacientů s FA mají v důsledku poruchy funkce uvedených genů nízkou toleranci vůči poškození DNA jak in vitro, tak in vivo.
Od prvního popisu molekulárně-genetického defektu u FA v roce 1992 bylo dodnes identifikováno přes 1 000 různých mutací 21 genů (FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCM, PALB2 (FANCN), RAD51C (FANCO), BRCA2, BRIP1, a SLX4 (FANCP), FANCQ, FANCV, FANCD1, FANCJ, FANCR, FANCS, FANCU. Největší skupinu mutací tvoří FANCA mutace, z ostatních jsou nejčastější FANCB a FANCC [30]. Asi 84 % pacientů je řazeno do subtypu A, C nebo G [5].
Z FA proteinů se jich 8 sdružuje v tzv. jaderný komplex a společně s FAN1 proteinem jsou nezbytné pro monoubikvitinaci FANCD2 proteinu. Porucha funkce každého z proteinů tohoto komplexu přerušují FANCD2 monoubikvitinaci, klíčový krok, který ovlivňuje translokaci jaderného komplexu k chromatinu a jeho asociaci s ostatními jadernými proteiny účastnící se buněčné odpovědi na poškození DNA (schéma).

FANCD1 gen je BCRA2 supresorový gen, poruchy jeho funkce vedou k narušení systému reparace DNA. FANCI gen kóduje helikázu, enzym který odvíjí DNA. FANCM gen ovlivňuje DNA translokaci, FANCL gen sdílí homologii s E3 ubikvitinovými ligázami.
FANCD2 gen je rovněž fosforylován ATM kinázou (ataxia-teleangiectasia protein) a ovlivňuje pak S-fázi buněčného cyklu se zástavou DNA syntézy při radiaci. Všechny tyto procesy hrají tedy důležitou roli v buněčné odpovědi na poškození DNA. Gen BRCA2 má souvislost s predispozicí ke vzniku karcinomu prsu a ovaria.
Diagnostika a léčba
U všech pacientů s anémií nebo trombocytopenií a typickými somatickými anomáliemi, u kterých vyslovíme podezření na FA, je indikováno provedení testů chromosomální fragility z lymfocytů periferní krve s použitím diepoxybutanu (DEB) nebo mitomycinu C (MMC). V případech chromosomálního mozaicizmu však tyto testy mohou prokázat pouze malé procento buněk s chromosomálními zlomy, průměrný počet zlomů potom může výsledně být normální. Proto u pacientů s podezřením na FA a negativním DEB testem se doporučuje doplnit vyšetření chromosomální nestability z fibroblastů periferní krve. Vzhledem ke znalosti kauzálních genů a k možnostem sekvenování nové generace je v dnešní době molekulárně-genetická diagnostika k jednoznačnému průkazu diagnózy lépe přístupná a snáze proveditelná.
Při plánování léčebné strategie je nezbytný komplexní multidisciplinární přístup, na kterém se kromě hematologů podílejí specialisté z oboru genetiky, onkologie, neurologie, kožního lékařství. Nedílnou součástí týmu by měl být i endokrinolog, protože u pacientů s FA se setkáváme s poruchami růstu, glukózového a lipidového metabolizmu, funkcí štítné žlázy, gonadálních funkcí a kostní denzity. Pacienti jsou hypersenzitivní k radiaci, kyslíkovým radikálům, a zejména k vlivu chemických mutagenů, proto musí být důsledně chráněni před jejich vlivem v běžném životě a zejména pak v případě nutnosti léčby maligních onemocnění.
Zcela nezbytné je pečlivé monitorování hematologických nálezů a periodické vyšetření kostní dřeně k vyloučení rozvoje MDS. K nepříznivému vývoji hematologického onemocnění může dojít velmi rychle. Proto je vhodné připravit rámcový plán transplantace kostní dřeně (HSCT). Vzhledem k radiosenzitivitě a chemosenzitivitě je nezbytné použít přípravné režimy s redukovanou intenzitou. Na funkci kostní dřeně, může mít u některých pacientů příznivý efekt léčba androgeny, na druhé straně však může mít nežádoucí účinky včetně zvýšeného rizika hepatoceluárního karcinomu.
K časnému odhalení karcinomů v oblasti hlavy, krku a genitálu je nezbytné periodické sledování ve specializovaných ambulancích, většinou 1krát za půl roku.
Lepším porozuměním genetické a molekulární podstatě onemocnění a zlepšením výsledků léčby se Fanconiho anémie v současné době mění na vzácné chronické onemocnění, které již zdaleka není doménou dětské hematologie, ale je diagnostikováno častěji i u dospělých a vyžaduje sofistikovaný multidisciplinární přístup. Je jedním z příkladů umožňujících hlubší porozumění interakcí genetické informace s prostředím v etiologii vrozených anomálií, hematopoézy a rozvoje maligního onemocnění.
Jsou vyvíjeny nové techniky prevence a léčby FA, např. HSCT od sourozence po výběru geneticky zdravého embrya, genová léčba, genomové editování s použitím genetické rekombinace nebo konstruovaných nukleáz. Tyto přístupy však jsou zatím v experimentální fázi.
prof. MUDr. Dagmar Pospíšilová, Ph.D.
Dětská klinika LF UP a FN Olomouc
Doručeno do redakce 12. 1. 2018
Přijato po recenzi 17. 3. 2018
Sources
- Pospíšilová D. Vrozené syndromy selhání kostní dřeně. Postgraduální medicína 2015; 17(6): 589–601.
- Vlachos A, Ball S, Dahl N et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol 2008; 142(6): 859–876. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1365–2141.2008.07269.x>.
- Vlachos A, Rosenberg PS, Atsidaftos E et al. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood 2012; 119(16): 3815–3819. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2011–08–375972>.
- Da Costa L, O’Donohue MF, van Dooijeweert B et al. Molecular approaches to diagnose Diamond-Blackfan anemia: The EuroDBA experience. Eur J Med Genet 2017; pii: S1769–7212(17)30505–0. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ejmg.2017.10.017>.
- Cmejlova J, Dolezalova L, Pospisilova D et al. Translational efficiency in patients with Diamond-Blackfan anemia. Haematologica 2006; 91(11): 1456–1464.
- Henter JI, Karlen J. Fatal agranulocytosis after deferiprone therapy in a child with Diamond-Blackfan anemia. Blood 2007; 109(12): 5157–5159. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2007–02–065805>.
- Pospisilova D, Cmejlova J, Hak J et al. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica 2007; 92(5): e66-e67.
- Narla A, Payne EM, Abayasekara N et al. L-Leucine improves the anaemia in models of Diamond Blackfan anaemia and the 5q - syndrome in a TP53-independent way. Br J Haematol 2014; 167(4): 524–528. Dostupné z DOI: <http://dx.doi.org/10.1111/bjh.13069>.
- Ear J, Huang H, Wilson T et al. RAP-011 improves erythropoiesis in zebrafish model of Diamond-Blackfan anemia through antagonizing lefty1. Blood 2015; 126(7): 880–890. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2015–01–622522>.
- Faivre L, Meerpohl J, Da Costa L et al. High-risk pregnancies in Diamond-Blackfan anemia: a survey of 64 pregnancies from the French and German registries. Haematologica 2006; 91(4): 530–533.
- Gambale A, Iolascon A, Andolfo I et al. Diagnosis and management of congenital dyserythropoietic anemias. Expert Rev Hematol 2016; 9(3): 283–296. Dostupné z DOI: <http://dx.doi.org/10.1586/17474086.2016.1131608>.
- Russo R, Gambale A, Langella C et al. Retrospective cohort study of 205 cases with congenital dyserythropoietic anemia type II: definition of clinical and molecular spectrum and identification of new diagnostic scores. Am J Hematol 2014; 89(14): E169-E175. Dostupné z DOI: <http://dx.doi.org/10.1002/ajh.23800>.
- Heimpel H, Kellermann K, Neuschwander N et al. The morphological diagnosis of congenital dyserythropoietic anemia: results of a quantitative analysis of peripheral blood and bone marrow cells. Haematologica 2010; 95(6): 1034–1036. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2009.014563>.
- Dgany O, Avidan N, Delaunay J et al. Congenital dyserythropoietic anemia type I is caused by mutations in codanin-1. Am J Hum Genet 2002; 71(6): 1467–1474. Dostupné z DOI: <http://dx.doi.org/10.1086/344781>.
- Babbs C, Roberts NA, Sanchez-Pulido L et al. Homozygous mutations in a predicted endonuclease are a novel cause of congenital dyserythropoietic anemia type I. Haematologica 2013; 98(9): 1383–1387. Dostupné z DOI: <http://dx.doi.org/10.3324/haematol.2013.089490>.
- Schwarz K, Iolascon A, Verissimo F et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet 2009; 41(8): 936–940.Dostupné z DOI: <http://dx.doi.org/10.1038/ng.405>.
- Irvine AF, Vikberg AL et al. Congenital dyserythropoietic anemia type III (CDA III) is caused by a mutation in kinesin family member, KIF23. Blood 2013; 121(23): 4791–4799. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2012–10–461392>.
- Sandström H, Wahlin A, Eriksson M et al. Intravascular haemolysis and increased prevalence of myeloma and monoclonal gammapathy in congenital dyserythropoietic anaemia, type III. Eur J Haematol 1994; 52(1): 42–46.
- Arnaud L, Saison C, Helias V et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet 2010; 87(5): 721–727. Dostupné z DOI: <http://dx.doi.org/10.1016/j.ajhg.2010.10.010>.
- Jaffray JA, Mitchell WB, Gnanapragasam MN et al. Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm. Blood Cells Mol Dis 2013; 51(2): 71–75. Dostupné z DOI: <http://dx.doi.org/10.1016/j.bcmd.2013.02.006>.
- Parez N, Dommergues M, Zupan V et al. Severe congenital dyserythropoietic anaemia type I: prenatal management, transfusion support and alpha-interferon therapy. Br J Haematol 2000; 110(2): 420–423.
- Lavabre-Bertrand T, Ramos J, Delfour C et al. Long-term alpha interferon treatment is effective on anaemia and significantly reduces iron overload in congenital dyserythropoiesis type I. Eur J Haematol 2004; 73(5): 380–383. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1600–0609.2004.00310.x>.
- Unal S, Russo R, Gumruk F et al. Successful hematopoietic stem cell transplantation in a patient with congenital dyserythropoietic anemia type II. Pediatr Transplant 2014; 18(4): E130-E133. Dostupné z DOI: <http://dx.doi.org/10.1111/petr.12254>.
- Ayas M, Al-Jefri A, Baothman A et al. Transfusion-dependent congenital dyserythropoietic anemia type I successfully treated with allogeneic stem cell transplantation. Bone Marrow Transplant 2002; 29(8): 681–682. Dostupné z DOI: <http://dx.doi.org/10.1038/sj.bmt.1703526>.
- Fanconi G. Familiäre, infantile, perniziosaartige Anämie (perniziöses Blutbiod and Konstitution). Jahrb Kinderheilk 1927; 117(2): 257–280.
- Verlander PC, Kaporis A, Liu Q et al. Carrier frequency of the IVS4 + 4 A-->T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood 1995; 86(11): 4034–4038.
- Neil VM, Fahmida E, Demuth I et al. A common Fanconi anemia mutation in black populations of sub-Saharan Africa. Blood 2005; 105(9): 3542–3544. Dostupné z DOI: <http://dx.doi.org/10.1182/blood-2004–10–3968>.
- Giampietro PF, Verlander PC, Davis JG et al. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genet 1997; 68(1): 58–61.
- Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev 2015; 33 : 32–40. Dostupné z DOI: <http://dx.doi.org/10.1016/j.gde.2015.07.002>.
- Cheung RS, Taniguchi. T Recent insights into the molecular basis of Fanconi anemia: genes, modifiers, and drivers. Int J Hematol 2017; 106(3): 335–344. Dostupné z DOI: <http://dx.doi.org/10.1007/s12185–017–2283–4>.
- Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Orkin SH, Ginsburg D, Look AT (eds). Nathan and Oski‘s Hematology of Infancy and Childhood. Vol 1, 6th ed. WB Saunders Philadelphia 2003 : 281–365. ISBN 978–1-4160–3430–8.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2018 Issue 5
Most read in this issue
- Autoimunitní hemolytická anémie
- Diferenciální diagnostika anémií
- Vrozené a získané krvácivé stavy
- Hemoglobinopatie