
Potenciál indukovaných pluripotentných kmeňových buniek v štúdiu a liečbe monogénového diabetu
Authors:
Terézia Valkovičová 1; Martina Škopková 1; Ying Cai 3; Juraj Staník 1,2; Miriam Cnop 3; Daniela Gašperíková 1
Authors‘ workplace:
DIABGENE & Ústav experimentálnej endokrinológie, Biomedicínske centrum SAV, Bratislava
1; Detská klinika LF UK a NÚDCH, Bratislava
2; ULB Center for Diabetes Research, Université Libre de Bruxelles, Belgicko
3
Published in:
Diab Obez 2021; 21(41): 7-15
Category:
Reviews
Overview
MEHMO a Wolframov syndróm sú z kategórie monogénových ochorení, u ktorých jedným z príznakov je aj neautoimunitný diabetes mellitus. Oba syndrómy sa zaraďujú medzi vzácne ochorenia. Získanie pacientskych B-buniek, na ktorých by bolo možné lepšie preštudovať mechanizmus vzniku ochorenia, je zložité až nemožné. Nové možnosti štúdia diabetu prináša metóda reprogramovania somatických buniek na bunky kmeňové a ich následná diferenciácia na B-bunky pankreasu. Takto pripravené B-bunky pankreasu by mohli byť využité nie len na vedecké štúdie, ale aj na bunkovú terapiu pacientov s monogénovým, prípadne polygénovým typom diabetu.
Klíčová slova:
ľudské indukované pluripotentné kmeňové bunky (hiPSC) – MEHMO – monogénový diabetes – Wolframov syndróm
Úvod
Monogénová forma diabetes mellitus. Monogénová forma diabetu je samostatná skupina v kategórii „špecifické typy diabetu“ podľa klasifikácie Americkej diabetologickej asociácie (ADA) [1]. Monogénový diabetes vzniká v dôsledku patologického variantu v jednom géne. Samotný klinický prejav závisí od patologického účinku danej mutácie. Pre monogénové formy diabetu je charakteristický rodinný výskyt ochorenia, prejavenie ochorenia v mladom veku a neprítomnosť autoimunitných protilátok (okrem prípadov monogénového autoimunitného diabetu), čím sa zároveň odlišujú od polygénových foriem diabetu [2].
Najaktuálnejšia klasifikácia podľa International Society for Pediatric and Adolescent Diabetes ISPAD [3], rozdeľuje typy monogénového diabetu u mladistvých na 4 kategórie:
neonatálny diabetes mellitus (nástup diabetu do 6 mesiacov od narodenia)
hyperglykémia alebo diabetes s rodinným výskytom a autozómovo-dominantným typom dedičnosti (Maturity Onset Diabetes of the Young – MODY)
genetické syndrómy asociované s diabetom (mitochondriálny diabetes, diabetes s renálnymi cystami, Wolframov syndróm)
monogénové syndrómy inzulínovej rezistencie (mutácie inzulínového receptora, lipoatrofický diabetes)
Epidemiológia monogénového diabetu nie je presne známa [4]. Najčastejším typom monogénového diabetu je hyperglykémia alebo diabetes, a to s rodinným výskytom a autozómovo dominantným typom dedičnosti, v literatúre nazývaný aj Maturity Onset Diabetes of the Young (MODY). Približne 1 % pacientov do 45 rokov, pôvodne klasifikovaných ako diabetes 1. typu (DM1T), a približne 4 % pacientov do 45 rokov, pôvodne klasifikovaných ako diabetes 2. typu (DM2T), sú v skutočnosti MODY-pacienti [5]. Druhý najčastejší typ monogénového diabetu je mitochondriálny diabetes (cca 0,5 % spomedzi všetkých diabetikov) [6]. Výskyt ostatných typov monogénového diabetu vrátane syndrómových foriem je pravdepodobne nižší.
Syndróm MEHMO
Medzi genetické syndrómy asociované s diabetom patrí aj syndróm MEHMO (Mental retardation, Epileptic seizures, Hypogonadism and Hypogenitalism, Microcephaly and Obesity). MEHMO je na X-chromozóm viazané recesívne ochorenie definované ako ťažká porucha intelektu v spojení s mikrocefáliou, epilepsiou, hypogonadizmom a hypogenitalizmom, diabetom a obezitou [7]. Syndróm vzniká v dôsledku mutácie v géne EIF2S3, ktorý kóduje podjednotku eukaryotického iniciačného faktora proteosyntézy γ (eIF2γ) potrebného pre iniciáciu translácie [8].
Wolframov syndróm
Ďalším zo syndrómov asociovaných s diabetom je Wolframov syndróm, známy tiež ako DIDMOAD (Diabetes Insipidus, early-onset Diabetes Mellitus, progressive Optic Atrophy, and Deafness). Klinické prejavy ochorenia sa zhoršujú vekom. Prvým klinickým prejavom býva spravidla diabetes mellitus s potrebnou inzulinoterapiou, ktorý nastupuje v prvej dekáde života (približne 6. rok života). Nasleduje atrofia zrakového nervu (približne 11. rok života), senzorineurálna porucha sluchu (približne 12,5 roka), centrálny diabetes insipidus (približne 14. rok života), dilatácia močových ciest až napokon neurologické abnormality [9,10]. Ochorenie je asociované s mutáciou v géne WFS1 kódujúcom wolframín, transmembránový proteín endoplazmatického retikula [11]. Vo väčšine prípadov ide o recesívne mutácie [12], no boli popísané aj heterozygotné a autosomálne dominantné mutácie, ktoré spôsobujú spektrum subfenotypov od izolovanej poruchy sluchu [13], izolovanej formy diabetu [14], kombináciu optickej atrofie a poruchy sluchu [15], až po klinicky závažný Wolframov synfróm s nástupom v neonatálnom veku [16].
Štúdium funkcie B-buniek
Zdokonalenie techník sekvenovania DNA prispelo k objavu mnohých monogénových ochorení, no napriek identifikovaným mutáciám v konkrétnych génoch, etiológia týchto ochorení nemusí byť známa. Wolframov syndróm je jedným z príkladov monogénových syndrómov, u ktorých prepojenie genotypu s fenotypom nie je úplne jasné [10,16]. Objasnenie etiológie monogénových syndrómov je do určitej miery jednoduchšie kvôli skutočnosti, že patogénna mutácia sa nachádza v jedinom géne. Monogénový diabetes tak môže predstavovať vhodný model pre štúdium mechanizmov poškodenia funkcie B-buniek pankreasu. Výsledky štúdia môžu byť prípadne aplikovateľné aj na polygénové typy diabetes mellitus.
Možnosti štúdia funkcie B-buniek v podmienkach in vivo sú limitované. Pre pochopenie patogénnych mechanizmov asociovaných s diabetom sú potrebné experimentálne modely buniek pankreasu. V súčasnosti existujú 3 možné spôsoby štúdia B-buniek: 1. primárne ostrovčeky izolované z pankreasu darcov orgánov, 2. humánne línie B-buniek a 3. bunky podobné ostrovčekom diferencované z ľudských pluripotentných kmeňových buniek (human Pluripotent Stem Cell – hPSC), ktoré zahŕňajú buď ľudské embryonálne (human Embyonic Stem Cell) alebo ľudské indukované pluripotentné kmeňové bunky (human-induced Pluripotent Stem Cell – hiPSCs).
Primárne ostrovčeky
Primárne ostrovčeky pankreasu sa získavajú od donorov pankreasu alebo počas operácie pankreasu, pričom ich veľkou výhodou je izolácia bez porušenia krvného toku, čím bunky v ostrovčekoch nestrácajú svoju funkčnosť [17]. Stanovenie transkripčného profilu týchto ostrovčekov spolu s GWAS (Genome Wide Association Studies) významne uľahčilo poznanie expresie génov, ako aj poznanie efektu génových variácií na expresiu génov. Toto umožnilo objav mnohých génov s potenciálnou úlohou v metabolizme glukózy a sekrécii inzulínu [18–22].
Stabilné bunkové línie
Ďalšou možnosťou ako študovať funkcie B-buniek je využitie stabilných bunkových línií, ktoré môžu byť živočíšneho alebo ľudského pôvodu. Vo veľkej miere sa používajú bunkové línie produkujúce inzulín, ktoré pochádzajú z myší, potkanov, či škrečkov, ako napr. RINm5F, HIT-T15, MIN-6, INS-1, B-TC (tab) [23–27]. Myšacie a potkanie bunkové modely prispeli k mnohým objavom v biológii B-buniek, sú pomerne stabilné, ich počet pasáží (t.j. počet prenesení buniek do novej kultivačnej nádoby) je menej limitovaný a celkové náklady na ich kultiváciu sú nižšie [17]. Endokrinné bunky pankreasu hlodavcov sa každopádne odlišujú od tých ľudských a existujú aj medzidruhové rozdiely v usporiadaní ich A - a B-buniek pankreasu. Medzičasom sa stali dostupnejšie práve ľudské bunkové línie, napr. EndoC-BH1 [27], ktoré jednoznačne prispievajú k ešte väčšiemu pochopeniu funkcie a fyziológie ľudských B-buniek, ako aj ich reakcie na rôzne liečivá [28–30]. Spomínané línie sú endokrinné a sú schopné produkovať a sekretovať inzulín po stimulácii glukózou. Každá z týchto bunkových línií má taktiež svoje limitácie (tab.).
![Najpoužívanejšie B-bunkové línie. Upravené podľa [79]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/fa5964c626f41970dbfa5d8ac9cd1de8.png)
V prípade štúdia patofyziológie B-buniek spôsobenej mutáciou v konkrétnom géne u pacientov s monogénovým diabetom majú spomínané bunkové modely značné limity. Nakoľko sa nejedná o bunky izolované od konkrétnych pacientov, nie sú schopné kopírovať fenotyp ich B-buniek. Naším cieľom bolo reprogramovať somatické bunky pacientov s MEHMO a Wolframovým syndrómom na indukované pluripotentné kmeňové bunky.
Indukované pluripotentné kmeňové bunky
Kvôli limitáciám B-bunkových stabilných línií sa stáva čoraz populárnejšou metóda vytvorenia indukovaných pluripotentných kmeňových buniek (iPSC). Ide o prípravu kmeňových buniek zo somatických buniek pacienta, ktoré nesú pacientovu kompletnú genetickú informáciu. Najčastejšie využívaným typom somatických buniek pre reprogramovanie sú fibroblasty, resp. PBMC-bunky (Peripheral Blood Mononucelar Cells), ktoré sú pomerne jednoducho získateľné.
Technika reprogramovania
Fusaki et al [31] vytvorili unikátnu metódu, pri ktorej sú somatické ľudské bunky reprogramované prostredníctvom špeciálne navrhnutého Sendai vírusového vektora (SeV) na pluripotentné kmeňové bunky. Vírusový vektor v sebe nesie zakódovanú informáciu pre markery ľudských kmeňových buniek (hESC, human Embryonic Stem-Cells): OCT3/4, SOX2, KLF4 a c-Myc. Tieto 4 transkripčné faktory sú nevyhnutné na spustenie reprogramovania buniek zo somatických na kmeňové. Jedná sa o kaskádovú reakciu, kedy expresia transkripčných faktorov „zapne“ ostatné signálne dráhy a expresiu génov zodpovedných za reprogramovanie diferencovanej bunky na kmeňovú bunku. Z vírusového vektora sú už na 3. deň od infikovania v bunkách exprimované endogénne markery pluripotencie Oct3/4, Sox2, Nanog, GDF3, TDGF1, Zfp42, Sal4F, Dnmt3b, CABRB3, CYP26A1, a FOXD3; telomerázová reverzná transkriptáza (hTERT) a povrchové markery pluripotencie SSEA4 a TRA-1–60 a -81. SeV vektor sa replikuje konštitutívne v cytoplazme infikovaných buniek, neintegruje sa do genómu a rastom a množením buniek sa z nich časom vymyje [31].
V súčasnosti sú publikované viaceré štúdie s iPSC bunkami od pacientov s monogénovým diabetom vrátane MEHMO [32] a Wolframového syndrómu [33–36]. V spolupráci s Université Libre de Bruxelles (ULB) Center for Diabetes Research sme reprogramovali PBMC bunky od pacienta so syndrómom MEHMO. V predchádzajúcich analýzach sme u pacienta identifikovali pomocou celoexómového sekvenovania (Whole Exome Sequencing – WES) variant c.1394_1397delTCAA v géne EIF2S3 spôsobujúci frameshift v eukaryotickom iniciačnom faktore eIF2γ s predčasným stop kodónom p.I465Sfs*4 [8]. Reprogramovali sme taktiež PBMC-bunky od pacienta s Wolframovým syndrómom, u ktorého sme pomocou Sangerovho sekvenovania identifikovali mutáciu c.2608_2619del v géne WFS1 spôsobujúcu deléciu 4 aminokyselín p.(870_873del) vo wolframíne.
Približne po 2 týždňoch kultivácie izolovaných PBMC-buniek s vírusovým vektorom začali bunky vytvárať prvé kolónie, ktoré niesli morfologické znaky ľudských kmeňových buniek. Kultiváciou a pasážovaním (prekultivovaním do novej kultivačnej nádoby) kolónií sme vypestovali iPSC-kolónie od pacienta s MEHMO syndrómom (obr. 1) a Wolframovým syndrómom. iPSC kolónie od pacienta s Wolframovým syndrómom však často podliehali spontánnej diferenciácii (obr. 2). Práve kvôli nestabilite iPSC-buniek je potrebné vykonať ich komplexnú kontrolu kvality.
Uverejnené so súhlasom autorky Y. Cai
a ULB Center for Diabetes Research,
Université Libre de Bruxelles,Belgicko

Uverejnené so súhlasom autorky T. Valkovičovej
a ULB Center for Diabetes Research, Université
Libre de Bruxelles,Belgicko
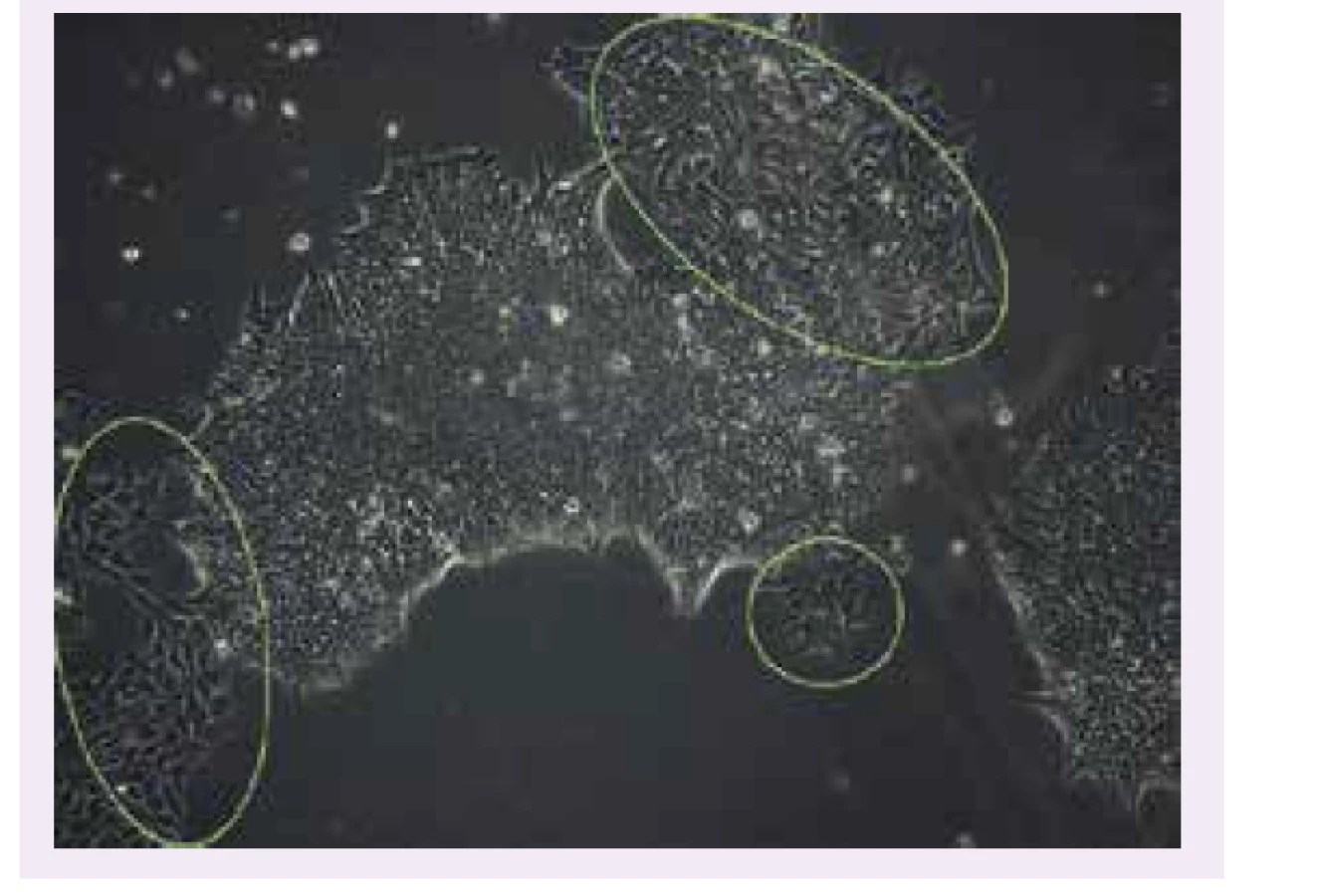
Kontrola kvality reprogramovania
Tak ako práca s ľudskými embryonálnymi kmeňovými bunkami a modelovými pankreatickými bunkovými líniami, aj práca s ľudskými indukovanými pluripotentnými kmeňovými bunkami a z nich diferencovanými B-bunkami má svoje limitácie. V súčasnosti sú stále diskutované jednotlivé kroky reprogramovania aj diferenciácie buniek [37,38]. iPSC-bunky môžu mať nestabilný genóm, nestabilitu pluripotencie, a to nie len medzi iPSC-líniami rôznych pacientov, ale aj medzi iPSC-klonmi v rámci jednej bunkovej línie. V tomto kontexte bolo publikovaných viacero štúdií, ktoré sa venovali nestabilite genómu vedúcich k diferenciácii ľudských kmeňových buniek na nádorové bunky [39–43]. Pri iPSC-bunkách je problémom taktiež spontánna diferenciácia. Riadená diferenciácia iPSC-buniek je častokrát neúspešná, nedostatočná, a opäť veľmi variabilná medzi jednotlivými bunkovými kolóniami či líniami [39,44–48].
Práve kvôli variabilite získaných iPSC sú potrebné rozsiahle kontrolné testy, v ktorých sa analyzuje ich pluripotencia, a to a) sledovaním expresie markerov pluripotencie pomocou imunocytologického farbenia a kvantitatívnej PCR; b) zistenie potenciálu diferencovať sa na 3 zárodočné vrstvy; c) kontrolou vymytia vírusového vektora použitého na reprogramovanie; d) analýzy karyotypu a genotypu pre prípadné odhalenie chromozomálnych aberácií alebo nechcene vnesených DNA-mutácií; e) v neposlednom rade je dôležitá kontrola čistoty, resp. prípadnej kontaminácie. Samotná príprava, reprogramovanie a kontrola iPSC trvá 3–6 mesiacov.
Vybrané iPSC-kolónie od pacientov s MEHMO a Wolframovým syndrómom sme podrobili kontrole kvality. qPCR potvrdila expresiu faktorov pluripotencie Nanog, Oct3/4, TNGF a Sox2. Imunocytochemické farbenie potvrdilo expresiu endogénnych faktorov pluripotencie Nanog a OCT3/4, ako aj povrchových markerov SSEA4, TRA-1–60. iPSC navyše preukázali potenciál diferencovať sa na 3 zárodočné vrstvy – endoderma, mezoderma, exoderma. Nezmenený karyotyp bol potvrdený G-prúžkovaním chromozómov, zachovanie genotypu bolo potvrdené prítomnosťou mutácie v géne EIF2S3 Sangerovým sekvenovaním. Vybraná iPSC-kolónia od MEHMO pacienta prešla kontrolou neprítomnosti bakteriálnej kontaminácie i vymytia vírusového vektora (pasáž 9).
Zvolený iPSC-klon od pacienta s Wolframovým syndrómom bol negatívny pre prítomnosť vírusového vektora (už v pasáži 7), imunocytochemické farbenie však ukázalo, že bunky boli síce pozitívne pre endogénne aj povrchové faktory pluripotencie, no v prítomnosti spontánne diferencovaných buniek, ktoré začali vytvárať svoje vlastné kolónie (obr. 3). Preto budú dodatočne zvolené iné iPSC-klony, ktoré podstúpia kontrolu kvality. Reprogramované bunky môžu byť zmrazené v médiu s DMEM (Dulbecco′s Modified Eagle′s – Medium), inaktivovaným Fetal Bovine Serum (FBS) a Dimethyl Sulfoxide (DMSO) v kryoskúmavkách pri -80°C a následne uskladnené v tekutom dusíku pri -196 °C.
Diferenciácia iPSC na pankreatické B-bunky
iPSC-bunky vďaka svojej pluripotentnosti môžu byť diferencované in vitro na viaceré typy buniek, ako napr. bunky krvi a endotelu [49], B-bunky [50], neuróny [51,52], či bunky očnej sietnice [53]. Z iPSC-buniek nie je možné vypestovať embryo či placentu. Diferenciáciou iPSC-buniek od pacienta na pankreatické B-bunky získavame unikátny model, na ktorom je možné študovať mechanizmus sekrécie inzulínu, reakciu bunky na príjem glukózy, bunkový stres, apoptózu atď. Prvý protokol pre diferenciáciu ľudských B-buniek z ľudských kmeňových buniek, ktoré boli použité na následnú transplantáciu, bol etablovaný v rokoch 2006 až 2008 [54,55].
Uverejnené so súhlasom autorky T. Valkovičovej a ULB Center for Diabetes Research, Université Libre de
Bruxelles,Belgicko

Na to, aby sa z iPSC-buniek diferencovalo dostatočné množstvo B-buniek, ktoré sú schopné sekretovať inzulín, je nevyhnutné, aby exprimovali 2 transkripčné faktory – PDX1 a NKX6.1. Tie sú prirodzene exprimované vo veľkej miere v progenitorových i maturovaných bunkách pankreasu [56]. In vitro diferencované B-bunky nie sú úplne maturované B-bunky pankreasu, ale B-like bunky, ktoré sú schopné úplne dozrieť až po transplantácii napr. do myšacej obličkovej kapsule [57,58], alebo do ľudskej pečene cez portálnu žilu [59,60]. Progenitorové bunky pankreasu ako aj B-bunky získané z ľudských embryonálnych kmeňových buniek sú po transplantácii schopné zvrátiť hyperglykémiu v imunodeficientných myšiach s DM1T a DM2T [57,61–63]. Aby sa v transplantovaných B-bunkách prejavila sekrécia inzulínu regulovaná glukózou, je potrebné, aby bunky dozrievali v tele dlhší čas [57,58,64–66].
Vďaka tomu majú aj z iPSC derivované B-bunky po ich transplantácii potenciál aspoň čiastočne a aspoň na určitú dobu nahradiť funkciu poškodených B-buniek pacienta. V súčasnosti sa zdokonaľujú najmä techniky transplantácie B-buniek odvodených od iPSC pacientom s diabetom [37,67–70].
Diskusia
Reprogramovanie somatických buniek pacientov s diabetom na indukované pluripotentné kmeňové bunky a ich následná diferenciácia predstavujú jedinečný model pre molekulárno-genetické štúdie mechanizmov ochorenia a štúdium účinku liečiv. Okrem štúdia narušených bunkových mechanizmov vedúcich k diabetu majú diferencované B-bunky veľký potenciál v bunkovej terapii. B-bunky diferencované in vitro síce nemajú sekrečnú kapacitu ako B-bunky izolované zo živého organizmu, no sú schopné reagovať na zvýšenú hladinu glukózy zvýšenou sekréciou inzulínu [62,71,72].
Indukované pluripotentné kmeňové bunky sa odlišujú od embryonálnych ľudských kmeňových buniek. Ak by boli bunky transplantované pacientovi, imunitný systém ich dokáže rozpoznať ako vlastné, a tým sa eliminuje riziko odmietnutia či nutnosť imunosupresívnej liečby. Na rozdiel od výskumu s ľudskými embryonálnymi bunkami, pri iPSC je do značnej mieri vyriešený etický problém práce s kmeňovými bunkami. iPSC-bunky prinášajú aj možnosť štúdia organogenézy, čo u diferencovaných buniek už možné nie je [73]. Je však dôležité poznamenať, že izolované bunky nemôžu poskytnúť presný obraz fungovania endokrinného pankreasu, pretože nie sú inervované a vaskularizované [28].
iPSC-kmeňové bunky sú v pomerne veľkej miere využívané pre účely úpravy genetickej informácie. Revolučná metóda DNA-editingu, Crispr-Cas9, umožňuje v živej bunke zaviesť špecifickú mutáciu na špecifické miesto v DNA, a tým vytvoriť model B-bunky pacienta s monogénovým diabetom [74–78], t. j. model „disease on a dish“. Takýmto spôsobom je možné získať vzácne pacientske B-bunky bez nutnosti biopsie pankreasu. Pomocou Cas9-endonukleázy, ktorá štiepi dvojvláknovú DNA, je mutácie možné nielen vniesť, ale aj odstrániť. Odstránením patogénnej mutácie v pacientových iPSC sa získava jedinečná izogénna kontrolná línia buniek, ktorá je adekvátnejšia ako kontrolné iPSC-bunky izolované z iného donora bez diabetu. „Opravené“ iPSC-bunky môžu byť následne diferencované na B-bunky pankreasu (schéma). V prípade syndrómových ochorení, akými sú aj MEHMO a Wolframov syndróm, by sa pacientom dalo pomôcť nie len s transplantáciou funkčných inzulín sekretujúcich B-buniek, ale aj neurónov, či buniek iných poškodených orgánov.
![Schéma 1 Schéma vytvorenia izogénnej kontrolnej línie iPSC a izogénnej diabetickej línie iPSC. Od
zdravého jedinca bez diabetu, resp. od pacienta s monogénovým diabetom sú izolované mononukleárne
bunky z periférnej krvi alebo kožné fibroblasty, ktoré sú následne pomocou RNA-vírusového
vektora reprogramované na iPSC-bunky. Pomocou metódy CRIPSR/Cas9 môže byť do DNA v iPSC
od zdravého jedinca vnesená patogénna mutácia, ktorú chceme študovať a sledovať jej účinky napr.
na vývoj, funkciu a sekrečnú schopnosť diferencovaných B-buniek. Naopak, pomocou CRISPR/Cas9
môžeme odstrániť patogénnu mutáciu z genómu iPSC-buniek od pacienta s monogénovým diabetom,
čím získame jedinečnú izogénnu kontrolnú líniu. Upravené podľa [80]](https://pl-master.mdcdn.cz/media/image_pdf/05a4b2071ecbe44830c068e688bf4e34.png?version=1622123380)
Reprogramovaniu a diferenciácii buniek in vitro sa venuje čoraz viac laboratórií. Indukované pluripotentné kmeňové bunky boli úspešne pripravené zo somatických buniek nie len od pacientov s MEHMO [32] a Wolframovým syndrómom [33,34], ale aj DM1T, DM2T, MODY1/2/3/5/8, či nediabetických nosičov mutácie v géne PDX1, ale aj zdravých kontrolných jedincov [68]. V rámci vedecko-výskumných aktivít laboratória Diabgene Ústavu experimentálnej endokrinológie Biomedicínskeho centra SAV v spolupráci s Université Libre de Bruxelles plánujeme diferenciáciu iPSC-buniek na pankreatické B-bunky, izolovaných z PBMC od pacienta s MEHMO syndrómom.
Napriek svojim obrovským výhodám sú iPSC a z nich derivované B-bunky stále systémom, ktorý nie je dokonalý. Reprogramovanie je proces, ktorý si vyžaduje týždne až mesiace, kým sa získajú stabilné pluripotentné kmeňové bunky. Je nevyhnutné, aby sa procesy reprogramovania a diferenciácie zjednotili a vylepšili na takú úroveň, aby prinášali stabilné bunkové línie, ktoré majú menšiu diverzitu v rámci kolónií. Zároveň je potrebné eliminovať technické nedostatky, ktoré môžu brzdiť napredovanie tejto metodiky. Pri diferenciácii na B-bunky bude obzvlášť dôležité vylepšiť „proteínový kokteil“, ktorý je bunkám pridávaný, aby sa dokázali cez jednotlivé štádiá postupne diferencovať z kmeňových buniek až na bunky produkujúce inzulín v závislosti od podanej koncentrácie glukózy in vitro.
Grantová podpora: Výskumný pobyt v Center for Diabetes Research, Université Libre de Bruxxelles (Brusel, Belgicko) bol podporený Slovenskou diabetologickou spoločnosťou; Národným štipendijným programom Slovenskej republiky a grantom APVV 170296.
Vyhlásenie o konflikte záujmov autora
Autor nevyhlasuje žiaden konflikt záujmov.
RNDr. Daniela Gašperíková, DrSc.
www.endo.sav.sk
Doručené do redakcie | Received 1. 3. 2021
Prijaté po recenzii | Accepted 29. 3. 2021
Sources
- [American Diabetes Association]. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019; 42(Suppl 1): S13-S28. Dostupné z DOI: <http://dx.doi.org/10.2337/dc19-S002].
- Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab 2008; 4(4): 200–213. Dostupné z DOI: <http://dx.doi.org/10.1038/ncpendmet0778>.
- Mayer-Davis EJ, Kahkoska AR, Jefferies C et al. ISPAD Clinical Practice Consensus Guidelines 2018: Definition, epidemiology, and classification of diabetes in children and adolescents. Pediatr Diabetes 2018; 19(Suppl 27): S7-S19. Dostupné z DOI: <http://dx.doi.org/10.1111/pedi.12773>.
- Hattersley A, Bruining J, Shield J et al. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 2009; 10(Suppl 12): S33-S42. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1399–5448.2009.00571.x>.
- Thanabalasingham G, Pal A, Selwood MP et al. Systematic assessment of etiology in adults with a clinical diagnosis of young-onset type 2 diabetes is a successful strategy for identifying maturity-onset diabetes of the young. Diabetes Care 2012; 35(6): 1206–1212. Dostupné z DOI: <http://dx.doi.org/10.2337/dc11–1243>.
- Murphy R, Turnbull DM, Walker M et al. Clinical features, diagnosis and management of maternally inherited diabetes and deafness (MIDD) associated with the 3243A>G mitochondrial point mutation. Diabet Med 2008; 25(4):383–399. Dostupné z DOI: <http://dx.doi.org/10.1111/j.1464–5491.2008.02359.x>.
- Steinmuller R, Steinberger D, Muller U. MEHMO (mental retardation, epileptic seizures, hypogonadism and -genitalism, microcephaly, obesity), a novel syndrome: assignment of disease locus to xp21.1-p22.13. Eur J Hum Genet 1998; 6(3): 201–206. Dostupné z DOI: <http://dx.doi.org/10.1038/sj.ejhg.5200180>.
- Skopkova M, Hennig F, Shin BS et al. EIF2S3 Mutations Associated with Severe X-Linked Intellectual Disability Syndrome MEHMO. Hum Mutat 2017; 38(4): 409–425. Dostupné z DOI: <http://dx.doi.org/10.1002/humu.23170>.
- Matsunaga K, Tanabe K, Inoue H et al. Wolfram syndrome in the Japanese population; molecular analysis of WFS1 gene and characterization of clinical features. PLoS One 2014; 9(9): e106906. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pone.0106906>.
- de Heredia ML, Cleries R, Nunes V. Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med 2013; 15(7): 497–506. Dostupné z DOI: <http://dx.doi.org/10.1038/gim.2012.180>.
- Hatanaka M, Tanabe K, Yanai A et al. Wolfram syndrome 1 gene (WFS1) product localizes to secretory granules and determines granule acidification in pancreatic beta-cells. Hum Mol Genet 2011; 20(7):1274–1284. Dostupné z DOI: <http://dx.doi.org/10.1093/hmg/ddq568>.
- Barrett TG, Poulton K, Bundey S. DIDMOAD syndrome; further studies and muscle biochemistry. J Inherit Metab Dis 1995; 18(2): 218–220. Dostupné z DOI: <http://dx.doi.org/10.1007/BF00711771>.
- Bespalova IN, Van Camp G, Bom SJ, et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum Mol Genet 2001; 10(22): 2501–2508. Dostupné z DOI: <http://dx.doi.org/10.1093/hmg/10.22.2501>.
- Bonnycastle LL, Chines PS, Hara T et al. Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes 2013; 62(11): 3943–3950. Dostupné z DOI: <http://dx.doi.org/10.2337/db13–0571>.
- Hogewind BF, Pennings RJ, Hol FA et al. Autosomal dominant optic neuropathy and sensorineual hearing loss associated with a novel mutation of WFS1. Mol Vis 2010; 16 : 26–35.
- De Franco E, Flanagan SE, Yagi T et al. Dominant ER Stress-Inducing WFS1 Mutations Underlie a Genetic Syndrome of Neonatal/Infancy-Onset Diabetes, Congenital Sensorineural Deafness, and Congenital Cataracts. Diabetes 2017; 66(7): 2044–2053. Dostupné z DOI: <http://dx.doi.org/10.2337/db16–1296>.
- Geraghty RJ, Capes-Davis A, Davis JM et al. Guidelines for the use of cell lines in biomedical research. Br J Cancer 2014; 111(6): 1021–1046. Dostupné z DOI: <http://dx.doi.org/10.1038/bjc.2014.166>.
- Taneera J, Fadista J, Ahlqvist E et al. Identification of novel genes for glucose metabolism based upon expression pattern in human islets and effect on insulin secretion and glycemia. Hum Mol Genet 2015; 24(7): 1945–1955. Dostupné z DOI: <http://dx.doi.org/10.1093/hmg/ddu610>.
- Fadista J, Vikman P, Laakso EO et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci U S A 2014; 111(38): 13924–13929. Dostupné z DOI: <http://dx.doi.org/10.1073/pnas.1402665111>.
- Pasquali L, Gaulton KJ, Rodriguez-Segui SA et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet 2014; 46(2): 136–143. Dostupné z DOI: <http://dx.doi.org/10.1038/ng.2870>.
- van de Bunt M, Manning Fox JE, Dai X et al. Transcript Expression Data from Human Islets Links Regulatory Signals from Genome-Wide Association Studies for Type 2 Diabetes and Glycemic Traits to Their Downstream Effectors. PLoS Genet 2015; 11(12): e1005694. Dostupné z DOI: <http://dx.doi.org/10.1371/journal.pgen.1005694>.
- Moran I, Akerman I, van de Bunt M et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab 2012; 16(4): 435–448. Dostupné z DOI: <http://dx.doi.org/10.1016/j.cmet.2012.08.010>.
- Gazdar AF, Carney DN, Russell EK et al. Establishment of continuous, clonable cultures of small-cell carcinoma of lung which have amine precursor uptake and decarboxylation cell properties. Cancer Res 1980; 40(10): 3502–3507.
- Santerre RF, Cook RA, Crisel RM et al. Insulin synthesis in a clonal cell line of simian virus 40-transformed hamster pancreatic beta cells. Proc Natl Acad Sci U S A 1981; 78(7): 4339–4343. Dostupné z DOI: <http://dx.doi.org/10.1073/pnas.78.7.4339>.
- Efrat S, Leiser M, Surana M et al. Murine insulinoma cell line with normal glucose-regulated insulin secretion. Diabetes 1993; 42(6): 901–907. Dostupné z DOI: <http://dx.doi.org/10.2337/diab.42.6.901>.
- Miyazaki J, Araki K, Yamato E et al. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 1990; 127(1): 126–132. Dostupné z DOI: <http://dx.doi.org/10.1210/endo-127–1-126>.
- Ravassard P, Hazhouz Y, Pechberty S et al. A genetically engineered human pancreatic beta cell line exhibiting glucose-inducible insulin secretion. J Clin Invest 2011; 121(9): 3589–3597. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI58447>.
- Carlessi R, Chen Y, Rowlands J et al. GLP-1 receptor signaling promotes beta-cell glucose metabolism via mTOR-dependent HIF-1alpha activation. Sci Rep 2017; 7(1): 2661. Dostupné z DOI: <http://dx.doi.org/10.1038/s41598–017–02838–2>.
- Ulrich AB, Schmied BM, Standop J et al. Pancreatic cell lines: a review. Pancreas 2002; 24(2): 111–120. Dostupné z DOI: <http://dx.doi.org/10.1097/00006676–200203000–00001>.
- Skelin M, Rupnik M, Cencic A. Pancreatic beta cell lines and their applications in diabetes mellitus research. ALTEX 2010; 27(2): 105–113. Dostupné z DOI: <http://dx.doi.org/10.14573/altex.2010.2.105>.
- Fusaki N, Ban H, Nishiyama A et al. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 2009; 85(8): 348–362. Dostupné z DOI: <http://dx.doi.org/10.2183/pjab.85.348>
- Young-Baird SK, Shin BS, Dever TE. MEHMO syndrome mutation EIF2S3-I259M impairs initiator Met-tRNAiMet binding to eukaryotic translation initiation factor eIF2. Nucleic Acids Res 2019; 47(2): 855–867. Dostupné z DOI: <http://dx.doi.org/10.1093/nar/gky1213>.
- Urano F. Wolfram syndrome iPS cells: the first human cell model of endoplasmic reticulum disease. Diabetes 2014; 63(3): 844–846. Dostupné z DOI: <http://dx.doi.org/10.2337/db13–1809>.
- Lu S, Kanekura K, Hara T et al. A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci U S A 2014; 111(49): E5292-E5301. Dostupné z DOI: <http://dx.doi.org/10.1073/pnas.1421055111>.
- Shang L, Hua H, Foo K et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014; 63(3): 923–933. Dostupné z DOI: <http://dx.doi.org/10.2337/db13–0717>.
- Grzela DP, Marciniak B, Pulaski L. Characterization of an induced pluripotent stem cell line (IMBPASi001-A) derived from fibroblasts of a patient affected by Wolfram Syndrome. Stem Cell Res 2020; 46 : 101858. Dostupné z DOI: <http://dx.doi.org/10.1016/j.scr.2020.101858>.
- Cierpka-Kmiec K, Wronska A, Kmiec Z. In vitro generation of pancreatic beta-cells for diabetes treatment. I. beta-like cells derived from human pluripotent stem cells. Folia Histochem Cytobiol 2019; 57(1): 1–14. Dostupné z DOI: <http://dx.doi.org/10.5603/FHC.a2019.0001>.
- Zhou Q, Melton DA. Pancreas regeneration. Nature 2018; 557(7705): 351–358. Dostupné z DOI: <http://dx.doi.org/10.1038/s41586–018–0088–0>.
- Laurent LC, Ulitsky I, Slavin I et al. Dynamic changes in the copy number of pluripotency and cell proliferation genes in human ESCs and iPSCs during reprogramming and time in culture. Cell Stem Cell 2011; 8(1): 106–118. Dostupné z DOI: <http://dx.doi.org/10.1016/j.stem.2010.12.003>.
- Imreh MP, Gertow K, Cedervall J et al. In vitro culture conditions favoring selection of chromosomal abnormalities in human ES cells. J Cell Biochem 2006; 99(2): 508–516. Dostupné z DOI: <http://dx.doi.org/10.1002/jcb.20897>.
- Baker DE, Harrison NJ, Maltby E et al. Adaptation to culture of human embryonic stem cells and oncogenesis in vivo. Nat Biotechnol 2007; 25(2): 207–215. Dostupné z DOI: <http://dx.doi.org/10.1038/nbt1285>.
- Maitra A, Arking DE, Shivapurkar N et al. Genomic alterations in cultured human embryonic stem cells. Nat Genet 2005; 37(10): 1099–1103. Dostupné z DOI: <http://dx.doi.org/10.1038/ng1631>.
- Mitalipova MM, Rao RR, Hoyer DM et al. Preserving the genetic integrity of human embryonic stem cells. Nat Biotechnol 2005; 23(1): 19–20. <http://dx.doi.org/10.1038/nbt0105–19>.
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature 2007; 448(7151): 313–317. <http://dx.doi.org/10.1038/nature05934>.
- Ramos-Mejia V, Munoz-Lopez M, Garcia-Perez JL et al. iPSC lines that do not silence the expression of the ectopic reprogramming factors may display enhanced propensity to genomic instability. Cell Res 2010; 20(10): 1092–1095. Dostupné z DOI: <http://dx.doi.org/10.1038/cr.2010.125>.
- Miura K, Okada Y, Aoi T et al. Variation in the safety of induced pluripotent stem cell lines. Nat Biotechnol 2009; 27(8): 743–745. Dostupné z DOI: <http://dx.doi.org/10.1038/nbt.1554>.
- Mayshar Y, Ben-David U, Lavon N et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell 2010; 7(4): 521–531. Dostupné z DOI: <http://dx.doi.org/10.1016/j.stem.2010.07.017>.
- Yamanaka S. Induced pluripotent stem cells: past, present, and future. Cell Stem Cell 2012; 10(6): 678–684. Dostupné z DOI: <http://dx.doi.org/10.1016/j.stem.2012.05.005>.
- Choi KD, Yu J, Smuga-Otto K et al. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells 2009; 27(3): 559–567. <http://dx.doi.org/10.1634/stemcells.2008–0922>.
- Demine S, Schiavo AA, Marin-Canas S et al. Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res Ther 2020; 11(1): 7. Dostupné z DOI: <http://dx.doi.org/10.1186/s13287–019–1523–3>.
- Hu BY, Weick JP, Yu J et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci U S A 2010; 107(9): 4335–4340. Dostupné z DOI: <http://dx.doi.org/10.1073/pnas.0910012107>.
- KarbalaeiMahdi A, Shahrousvand M, Javadi HR et al. Neural differentiation of human induced pluripotent stem cells on polycaprolactone/gelatin bi-electrospun nanofibers. Mater Sci Eng C Mater Biol Appl 2017; 78 : 1195–1202. <http://dx.doi.org/10.1016/j.msec.2017.04.083>.
- Chichagova V, Hilgen G, Ghareeb A et al. Human iPSC differentiation to retinal organoids in response to IGF1 and BMP4 activation is line - and method-dependent. Stem Cells 2020; 38(2): 195–201. <http://dx.doi.org/10.1002/stem.3116>.
- Kroon E, Martinson LA, Kadoya K et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat Biotechnol 2008; 26(4): 443–452. Dostupné z DOI: <http://dx.doi.org/10.1038/nbt1393>.
- D’Amour KA, Bang AG, Eliazer S et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 2006; 24(11): 1392–1401. Dostupné z DOI: <http://dx.doi.org/10.1038/nbt1259>.
- Lyttle BM, Li J, Krishnamurthy M et al. Transcription factor expression in the developing human fetal endocrine pancreas. Diabetologia 2008; 51(7): 1169–1180. Dostupné z DOI: <http://dx.doi.org/10.1007/s00125–008–1006-z>.
- Rezania A, Bruin JE, Xu J et al. Enrichment of human embryonic stem cell-derived NKX6.1-expressing pancreatic progenitor cells accelerates the maturation of insulin-secreting cells in vivo. Stem Cells 2013; 31(11): 2432–2442. Dostupné z DOI: <http://dx.doi.org/10.1002/stem.1489>.
- Rezania A, Bruin JE, Riedel MJ et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012; 61(8): 2016–2029. Dostupné z DOI: <http://dx.doi.org/10.2337/db11–1711>.
- Onaca N, Naziruddin B, Matsumoto S et al. Pancreatic islet cell transplantation: update and new developments. Nutr Clin Pract 2007; 22(5): 485–493. Dostupné z DOI: <http://dx.doi.org/10.1177/0115426507022005485>.
- Robertson RP. Islet transplantation for type 1 diabetes, 2015: what have we learned from alloislet and autoislet successes? Diabetes Care 2015; 38(6): 1030–1035. Dostupné z DOI: <http://dx.doi.org/10.2337/dc15–0079>.
- Bruin JE, Saber N, Braun N et al. Treating diet-induced diabetes and obesity with human embryonic stem cell-derived pancreatic progenitor cells and antidiabetic drugs. Stem Cell Reports 2015; 4(4): 605–620. Dostupné z DOI: <http://dx.doi.org/10.1016/j.stemcr.2015.02.011>.
- Rezania A, Bruin JE, Arora P et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol 2014; 32(11): 1121–1133. Dostupné z DOI: <http://dx.doi.org/10.1038/nbt.3033>.
- Jeon K, Lim H, Kim JH et al. Differentiation and transplantation of functional pancreatic beta cells generated from induced pluripotent stem cells derived from a type 1 diabetes mouse model. Stem Cells Dev 2012; 21(14): 2642–2655. Dostupné z DOI: <http://dx.doi.org/10.1089/scd.2011.0665>.
- Xie R, Everett LJ, Lim HW et al. Dynamic chromatin remodeling mediated by polycomb proteins orchestrates pancreatic differentiation of human embryonic stem cells. Cell Stem Cell 2013; 12(2): 224–237. <http://dx.doi.org/10.1016/j.stem.2012.11.023>.
- Kelly OG, Chan MY, Martinson LA et al. Cell-surface markers for the isolation of pancreatic cell types derived from human embryonic stem cells. Nat Biotechnol 2011; 29(8): 750–756. Dostupné z DOI: <http://dx.doi.org/10.1038/nbt.1931>.
- van der Torren CR, Zaldumbide A, Duinkerken G et al. Immunogenicity of human embryonic stem cell-derived beta cells. Diabetologia 2017; 60(1): 126–133. Dostupné z DOI: <http://dx.doi.org/10.1007/s00125–016–4125-y>.
- Dinnyes A, Schnur A, Muenthaisong S et al. Integration of nano - and biotechnology for beta-cell and islet transplantation in type-1 diabetes treatment. Cell Prolif 2020; 53(5): e12785. Dostupné z DOI: <http://dx.doi.org/10.1111/cpr.12785>.
- Kondo Y, Toyoda T, Inagaki N et al. iPSC technology-based regenerative therapy for diabetes. J Diabetes Investig 2018; 9(2): 234–243. Dostupné z DOI: <http://dx.doi.org/10.1111/jdi.12702>.
- Lysy PA. [Cellular therapy of diabetes: focus on the latest developments]. Med Sci (Paris) 2016; 32(4): 401–407. Dostupné z DOI: <http://dx.doi.org/10.1051/medsci/20163204019>.
- Barkai U, Rotem A, de Vos P. Survival of encapsulated islets: More than a membrane story. World J Transplant 2016; 6(1): 69–90. Dostupné z DOI: <http://dx.doi.org/10.5500/wjt.v6.i1.69>.
- Kunisada Y, Tsubooka-Yamazoe N, Shoji M et al. Small molecules induce efficient differentiation into insulin-producing cells from human induced pluripotent stem cells. Stem Cell Res 2012; 8(2): 274–284. Dostupné z DOI: <http://dx.doi.org/10.1016/j.scr.2011.10.002>.
- Pagliuca FW, Millman JR, Gurtler M et al. Generation of functional human pancreatic beta cells in vitro. Cell 2014; 159(2): 428–439. Dostupné z DOI: <http://dx.doi.org/10.1016/j.cell.2014.09.040>.
- Suchy F, Yamaguchi T, Nakauchi H. iPSC-Derived Organs In Vivo: Challenges and Promise. Cell Stem Cell 2018; 22(1): 21–24. Dostupné z DOI: <http://dx.doi.org/10.1016/j.stem.2017.12.003>.
- Zhu Z, Li QV, Lee K et al. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell 2016; 18(6): 755–768. <http://dx.doi.org/10.1016/j.stem.2016.03.015>.
- McGrath PS, Watson CL, Ingram C et al. The Basic Helix-Loop-Helix Transcription Factor NEUROG3 Is Required for Development of the Human Endocrine Pancreas. Diabetes 2015; 64(7): 2497–2505. Dostupné z DOI: <http://dx.doi.org/10.2337/db14–1412>.
- Tiyaboonchai A, Cardenas-Diaz FL, Ying L et al. GATA6 Plays an Important Role in the Induction of Human Definitive Endoderm, Development of the Pancreas, and Functionality of Pancreatic beta Cells. Stem Cell Reports 2017; 8(3): 589–604. Dostupné z DOI: <http://dx.doi.org/10.1016/j.stemcr.2016.12.026>.
- Saarimaki-Vire J, Balboa D, Russell MA et al. An Activating STAT3 Mutation Causes Neonatal Diabetes through Premature Induction of Pancreatic Differentiation. Cell Rep 2017; 19(2): 281–294. Dostupné z DOI: <http://dx.doi.org/10.1016/j.celrep.2017.03.055>.
- Balboa D, Saarimaki-Vire J, Borshagovski D et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. Elife 2018; 7: e38519. Dostupné z DOI: <http://dx.doi.org/10.7554/eLife.38519>.
- Green AD, Vasu S, Flatt PR. Cellular models for beta-cell function and diabetes gene therapy. Acta Physiol (Oxf) 2018; 222(3). <http://dx.doi.org/10.1111/apha.13012>.
- Informácie dostupné z WWW: <https://smart.servier.com>.
Labels
Diabetology ObesitologyArticle was published in
Diabetes and obesity

2021 Issue 41
Most read in this issue
- Nesúlad medzi glykémiami a HbA1c – je možné vysvetlenie podľa biokinetického modelu glykácie?
- Potenciál indukovaných pluripotentných kmeňových buniek v štúdiu a liečbe monogénového diabetu
- Vieme dnes ovplyvniť nadváhu a obezitu – a tým predísť ich dôsledkom ako býva vývoj diabetu či kardiovaskulárneho ochorenia
- Novinky z oblasti bazálnych inzulínov 2. generácie pri diabete 1. typu a dáta z kontinuálneho monitorovania glykémie