
Aminopyrazinoic acid esters as potential antimycobacterial drugs
Authors:
Martin Doležal; Jana Vobicková; Barbora Servusová; Pavla Paterová
Authors‘ workplace:
Department of Clinical Microbiology, University Hospital, Hradec Králové, Czech Republic
; Charles University in Prague, Faculty of Pharmacy, Department of Pharmaceutical Chemistry and Drug Control
Published in:
Čes. slov. Farm., 2013; 62, 84-88
Category:
Original Articles
Overview
A series of esters of the 3-aminopyrazine-2-carboxylic acid as potential antimycobacterial drugs was synthesized. A CEM Discover microwave reactor with an autosampler Explorer 24 which served to accelerate the reaction was used for synthesis. The prepared products were characterized by IR, 1H NMR, 13C NMR spectra, elementary analysis and melting points. Log P and ClogP values were calculated. Final products were evaluated in vitro for their antimycobacterial activity. The most active compound was hexyl 3-aminopyrazine-2-carboxylate (7), whose antimycobacterial activity (MIC) against M. tuberculosis H37Rv was 6.25 μg/mL.
Keywords:
prodrugs – pyrazinoic acid esters – lipophilicity calculation – in vitro antimycobacterial activity
Introduction
Tuberculosis (TB), the world’s leading infectious disease, is caused by Mycobacterium tuberculosis, which is one of the most successful human pathogens. Treatment of TB requires an extra-long duration time using multiple drugs, divided into the first-line (isoniazid, rifampicin, pyrazinamide, ethambutol) and second line drugs. There is also an alarming emergence of multidrug resistant M. tuberculosis. As a result a need has arisen to develop novel anti-tubercular agents1). Pyrazinamide (PZA) and pyrazinoic acid (POA) as potential analeptic drugs have been synthesised in Germany2) and later in the USA as an intermediate compound on the pathway of aminopyrazine synthesis3), but the antimycobacterial activity of PZA was reported later, in 19524–8). It was soon shown that a mycobacterial enzyme called nicotinamidase hydrolyzed nicotinamide and pyrazinamide to the corresponding carboxylic acid, which is the actual active compound (see Fig. 1)9). PZA plays a unique role in modern TB chemotherapy10). Inclusion of PZA enables considerable shortening of the treatment period from the previously 9–12 months to 6 months, thus the drug plays a pivotal role in the current short-course chemotherapy for drug susceptible strains of TB. The powerful sterilizing activity of PZA is due to its ability to kill a population of persistent tubercle bacilli that are not killed by other TB drugs11). Furthermore, the synergistic activity of PZA with newly developed agents such as the diarylquinoline bedaquiline suggests that the use of PZA in regimens including novel agents could substantially improve efficacy, if the organism retains susceptibility to PZA12, 13).

Substituted pyrazinecarboxylic acid esters have previously shown a very good in vitro activity against M. avium and M. kansasii as well as against M. tuberculosis14). Pyrazinoic acid esters exhibit antimycobacterial activity probably due to better penetration through the mycobacterial cell wall, where they could be metabolized by mycobacterial esterases to the corresponding acid. While POA cannot pass through the mycobacterial cell walls due to its low lipophilicity, the concept of the POA prodrug is a suitable approach to increase the likelihood of its penetration into the resistant mycobacteria15, 16). Cynamon and colleagues15, 17) synthesized a series of un - or substituted POA (pyrazinecarboxylic, 5-fluoropyrazine-2-carboxylic, 5-methylpyrazine-2-carboxylic, and 5-chloropyrazine-2-carboxylic acid) esters, exhibiting a good in vitro antimycobacterial activity against several mycobacterial strains, including the PZA-resistant strain of M. tuberculosis16–26). A classical quantitative structure-activity relationships model for the selected compounds was proposed18, 19). Some 5-hydroxypyrazine-2-carboxylic acid derivatives are up to 1000-fold more active in vitro against M. tuberculosis and other Mycobacterium strains than existing antituberculous agents18); therefore substituted analogues of the POA prodrug with isosteric replacements of the hydroxylic moiety with the amino group were projected and prepared. The aim of our project was to develop some potential antimycobacterial prodrugs which can deliver the active agent to the site of the therapeutic action, to increase the drug bio-availability and selectivity of the therapeutic action and to present our results in the finding of new antimycobacterial active compounds based on esters 1–7 of 3-aminopyrazine-2-carboxylic acid (Fig. 2)27), i.e. prodrugs which can be easily activated by mycobacterial esterases.

Two series of aliphatic alcohols were chosen for the synthesis of final structures 1–7, namely four unbranched alcohols in the range from ethanol to n-hexanol and also three branched aliphatic alcohols – isopropanol, isobutanol and isopentanol.
Experimental part
Materials and methods
All organic solvents used for the synthesis were of analytical grade. All chemicals were purchased from Sigma-Aldrich (Schnelldorf, Germany). Compounds were synthesized using a microwave reactor CEM Discover with an autosampler Explorer 24 (CEM Corporation, Metthews, NC, USA). The reactions were monitored using Merck Silica 60 F254 TLC plates (Merck, Darmstadt, Germany). Compounds were purified using an automated chromatograph CombiFlash Rf (Teledyne Isco, Lincoln, NE, USA) using columns filled with Kieselgel 60, 0.040–0.063 mm (Merck, Darmstadt, Germany); gradient elution (hexane/ethyl-acetate), detection wavelength 260 nm, monitor wavelength 280 nm. NMR analysis was performed on a spectrometer Varian Mercury-Vx BB 500 (Varian, Palo Alto, CA, USA) at 500 MHz for 1H and 125 MHz for 13C. Chemical shifts were recorded as δ values in parts per million (ppm) and were indirectly referenced to tetramethylsilane (TMS). IR spectra were recorded in KBr blocks on a Nicolet Impact 400 (Nicolet, Madison, WI, USA). Elementary analysis was performed on a CE Instruments EA-1110 CHN analyser (CE Instruments, Wigan, UK). Melting points were determined on a Stuart SMP30 melting point apparatus (Bibby Scientific Limited, Staffordshire, UK) and are uncorrected.
Synthesis of 3-aminopyrazine-2-carboxylates
Final compounds were prepared via esterification of 3-aminopyrazine-2-carboxylic acid (see Fig. 2). Special thick-walled tubes intended for use in a microwave reactor were filled with a mixture of 3-aminopyrazine-2-carboxylic acid (3 mmol), appropriate alcohol (4 mL, used in excess and as a solvent) and 95–97.5% sulphuric acid (1.9 mmol). Tubes fitted with a stirrer and closed with a special cap were inserted into the reactor. Reaction conditions for synthesis of individual compounds are listed in Table 1.

The reaction was monitored using a TLC with hexane/ethyl acetate 1 : 2 mixture as the eluent. Then the solution was evaporated till dryness with sea sand and purified using a flash column chromatography (40 g column, gradient elution hexane/ethyl acetate). The summary of prepared derivatives and their physico-chemical data are listed in Tables 1 and 2.

Data of prepared compounds 1–7
Ethyl 3-aminopyrazine-2-carboxylate (1). White crystalline compound. M.p. 199.9–202.4 °C (ref. 28) gives m.p. 189–191 °C); 1H NMR (DMSO-d6) δ 8.24 (d, 1H, J=2.2 Hz, H5), 7.88 (d, 1H, J=2.2 Hz, H6), 7.37 (bs, 2H, NH2), 4.21 (q, 2H, J=7.1 Hz, OCH2), 1.09 (t, 3H, J=7.1 Hz, CH3); 13C NMR (DMSO-d6) δ 166.3, 156.1, 147.9, 132.6, 123.5, 61.1, 14.3; IR [cm-1] 3452 (-NH2), 2987, 1693 (C=O), 1604, 1304, 1190, 1106 (C-O), 815; Anal. Calcd. 50.29 % C, 5.43 % H, 25.14 % N; Found: 50.41 % C, 5.57 % H, 25.23 % N.
Isopropyl 3-aminopyrazine-2-carboxylate (2). White crystalline compound. M.p. 77.5–79.1 °C; 1H NMR (DMSO-d6) δ 8.24 (d, 1H, J=2.0 Hz, H5), 7.89 (d, 1H, J=2.0 Hz, H6), 7.32 (bs, 2H, NH2), 5.19–5.09 (m, 1H, OCH2), 1.30 (d, 6H, J=6.4 Hz, CH3); 13C NMR (DMSO-d6) δ 165.8, 156.1, 147.8, 132.6, 123.8, 68.7, 21.8; IR [cm-1] 3456 (-NH2), 2984, 2349, 1688 (C=O), 1604, 1302, 1187, 1091 (C-O), 816; Anal. Calcd. 53.03 % C, 6.12 % H, 23.19 % N; Found: 53.20 % C, 6.08 % H, 23.22 % N.
Butyl 3-aminopyrazine-2-carboxylate (3). White crystalline compound. M.p. 67.8–70.2 °C; 1H NMR (DMSO-d6) δ 8.25 (d, 1H, J=2.2 Hz, H5), 7.90 (d, 1H, J=2.2 Hz, H6), 7.32 (bs, 2H, NH2), 4.26 (t, 2H, J=6.7 Hz, OCH2), 1.72–1.62 (m, 2H, CH2), 1.43–1.33 (m, 2H, CH2), 0.91 (t, 3H, J=7.3 Hz, CH3); 13C NMR (DMSO-d6) δ 166.3, 156.1, 147.9, 132.7, 123.5, 64.7, 30.3, 18.9, 13.8; IR [cm-1] 3439 (-NH2), 2963, 2349, 1694 (C=O), 1603, 1309, 1192, 1104 (C-O), 816; Anal. Calcd. 55.37 % C, 6.71 % H, 21.52 % N; Found: 55.47 % C, 6.80 % H, 21.48 % N.
Isobutyl 3-aminopyrazine-2-carboxylate (4). White crystalline compound. M.p. 84.9–87.5 °C; 1H NMR (DMSO-d6) δ 8.18 (d, 1H, J=2.0 Hz, H5), 8.03 (d, 1H, J=2.0 Hz, H6), 7.26 (bs, 2H, NH2), 4.19 (d, 2H, J=6.9 Hz, OCH2), 2.06–1.95 (m, 1H, CH), 0.94 (d, 6H, J=6.6 Hz, CH3); 13C NMR (DMSO-d6) δ 166.3, 156.1, 147.9, 132.7, 123.5, 70.8, 27.5, 19.1; IR [cm-1] 3450 (-NH2), 2966, 1693 (C=O), 1609, 1310, 1194, 1095 (C-O), 814; Anal. Calcd. 55.37 % C, 6.71 % H, 21.52 % N; Found: 55.49 % C, 6.83 % H, 21.39 % N.
Pentyl 3-aminopyrazine-2-carboxylate (5). White crystalline compound. M.p. 62,7–65,0 °C; 1H NMR (DMSO-d6) δ 8.24 (d, 1H, J=2.2 Hz, H5), 7.89 (d, 1H, J=2.2 Hz, H6), 7.32 (bs, 2H, NH2), 4.25 (t, 2H, J=6.7 Hz, OCH2), 1.73–1.64 (m, 2H, CH2), 1.38–1.26 (m, 4H, CH2), 0.87 (t, 3H, J=7.1 Hz, CH3); 13C NMR (DMSO-d6) δ 166.3, 156.1, 147.9, 132.7, 123.5, 65.0, 28.0, 27.8, 21.9, 14.0; IR [cm-1] 3453 (-NH2), 2960, 2348, 1692 (C=O), 1606, 1300, 1192, 1102 (C-O), 816; Anal. Calcd. 57.40 % C, 7.23 % H, 20.08 % N; Found: 57.52 % C, 7.26 % H, 20.01 % N.
Isopentyl 3-aminopyrazine-2-carboxylate (6). White crystalline compound. M.p. 79,2–84,6 °C; 1H NMR (DMSO-d6) δ 8.24 (d, 1H, J=1.2 Hz, H5), 7.90 (d, 1H, J=1.2 Hz, H6), 7.32 (bs, 2H, NH2), 4.29 (t, 2H, J=6.7 Hz, OCH2), 1.73–1.60 (m, 1H, CH), 1.58 (q, 2H, J= 6.8 Hz, CH2), 0.91 (d, 3H, J=6.6 Hz, CH3); 13C NMR (DMSO-d6) δ 166.3, 156.1, 147.9, 132.7, 123.5, 63.5, 36.9, 24.7, 22.5; IR [cm-1] 3453 (-NH2), 2961, 2347, 1690 (C=O), 1605, 1300, 1187, 1098 (C-O), 816; Anal. Calcd. 57.40 % C, 7.23 % H, 20.08 % N; Found: 57.43 % C, 7.14 % H, 20.07 % N.
Hexyl 3-aminopyrazine-2-carboxylate (7). White crystalline compound. M.p. 77,6–79,8 °C; 1H NMR (DMSO-d6) δ 8.24 (d, 1H, J=2.2 Hz, H5), 7.90 (d, 1H, J=2.2 Hz, H6), 7.32 (s, 2H, NH2), 4.25 (t, 2H, J=6.6 Hz, OCH2), 1.72–1.63 (m, 2H, CH2), 1.40–1.19 (m, 6H, CH2), 0.85 (t, 3H, J=7.1 Hz, CH3); 13C NMR (DMSO-d6) δ 166.3, 156.1, 147.9, 132.7, 123.5, 65.1, 31.1, 28.3, 25.3, 22.2, 14.1; IR [cm-1] 3445 (-NH2), 2954, 2349, 1692 (C=O), 1600, 1310, 1189, 1105 (C-O), 817; Anal. Calcd. 59.17 % C, 7.67 % H, 18.82 % N; Found: 59.24 % C, 7.79 % H, 18.86 % N.
Lipophilicity calculation
Log P (the logarithm of the partition coefficient for n-octanol/water) and ClogP (the logarithm of n-octanol/water partition coefficient P based on established chemical interactions) were calculated using the program CS ChemBioDraw Ultra ver. 12.0 (CambridgeSoft, Cambridge, MA, USA). The results are shown in Table 2.
In vitro antimycobacterial activity
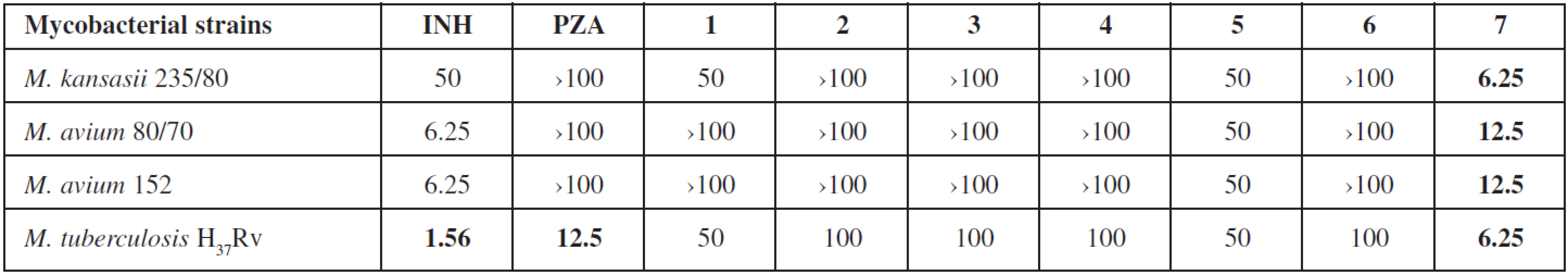
Antimycobacterial evaluation using the microdilution panel method was shielded by the Department of Clinical Microbiology, University Hospital and Faculty of Medicine in Hradec Králové, Charles University in Prague, Czech Republic. Four mycobacterial strains were used: M. tuberculosis H37Rv CNCTC My 331/88, M. avium CNCTC My 80/72, M. avium CNCTC My 152/73 and M. kansasii CNCTC My 235/80 (Czech National Collection of Type Cultures, National Institute of Public Health, Prague, Czech Republic). Tested compounds were dissolved in DMSO (to final concentrations 100, 50, 25, 12.5, 6.25, 3.125 and 1.563 μg/mL), diluted with Šula’s semisynthetic medium (Trios, Prague, Czech Republic) and placed into the microdilution panel. Tested species were added in the form of a suspension in an isotonic saline solution. The final concentration of DMSO did not exceed 1% (v/v), this concentration of DMSO did not affect the growth of mycobacteria. The cultures were grown in Šula’s semisynthetic medium at pH 5.7 and 37 °C. The antimycobacterial activity was determined visually after 14 days (6 days for M. kansasii) of incubation as the minimal inhibition concentration (MIC, μg/mL), i.e., the lowest concentration of the tested substance which inhibited the growth of mycobacteria.
Results and discussion
We decided to test whether the isosteric replacements of the hydroxylic moiety with the amino group approach could be used to improve the activity of POA ester. Our results are in good correlation with similar series published in 200929). However, there is some limitation to the use of such esters as drugs; efficacy studies in mice failed to show any antitubercular activity likely due to poor stability of the esters in plasma10). But another series of more lipophilic ester prodrugs (i.e. tetradecyl ester) were found to be active in concentrations 10-fold lower than those needed for PZA to kill sensitive M. tuberculosis and also have suitable stability in the presence of plasma29). 3-Aminopyrazine-2-carboxylates 1–7 have been synthesized using a microwave reactor and evaluated regarding to their antimycobacterial activity; six of them (2–7) are new compounds, ethyl 3-aminopyrazine-2-carboxylate (1) has been prepared by Vontor28) via alkoxycarbonylation of aminopyrazine. The esterification was an easy and quick process, but title esters were obtained by microwave assisted reaction in low yields (3.3–9.5%). We suppose that the main reasons are low stability in solution and also decarboxylation of 3-aminopyrazine-2-carboxylic acid. 2-Aminopyrazine, the main side product, was detected by 1H a 13C NMR spectra. We also planned to prepare and evaluate propyl ester of the starting compound, but the yields were too low and we were not successful to isolate it from the reaction mixture.
Lipophilicity
Lipophilicity, one of the most important physicochemical properties of the compound, which seems to be a key factor related to the cell transmembrane transport and other biological processes, can either be determined experimentally or predicted by means of the commercially available programmes. Log P/ClogP values of compounds 1–7 were calculated using the program ChemBioDraw Ultra (ver. 12.0) and the results are shown in Table 3. The ClogP value is correlated directly to the molecular hydrophobicity, and, thereby, to the diffusion through the biological membranes, i.e. into the mycobacterial cell wall. The lowest lipophilicity was shown by ethyl 3-aminopyrazine-2-carboxylate (1), while hexyl 3-aminopyrazine-2-carboxylate (7) was the most lipophilic compound of this series. Based on log P values, lipophilicity increased with the extension of the alkyl chain. This can be easily understood because mycolic acids in the cell wall provide high hydrophobicity to the mycobacteria, and then, highly hydrophobic compounds can cross the cell wall more easily19).
In vitro antimycobacterial evaluation
3-Aminopyrazine-2-carboxylate esters 1–7 have been evaluated regarding to their antimycobacterial activity against M. tuberculosis and several Mycobacteria Other Than Tuberculosis (MOTTs) (see Table 3). Only hexyl 3-aminopyrazine-2-carboxylate (7) possessed activity against M. tuberculosis H37Rv (MIC = 6.25 μg/mL) which was slightly better than MIC for PZA. More importantly, this compound also showed activity against the MOTTs tested, which are naturally unsusceptible to PZA. Other compounds did not exhibit any interesting activity against the tested strains. The obtained results provide some insight into the SAR in this series. The activity is probably lipophilicity dependent and culminates in the compound with hexyl substitution.
In vitro antibacterial and antifungal evaluation
All prepared compounds were tested for their in vitro antibacterial30, 31) and antifungal32) activity. None of the synthesized compounds exhibited any activity against the strains tested.
Conclusion
The present study has shown that long chain esters of 3-aminopyrazine-2-carboxylic acid possess better antimycobacterial properties and that higher lipophilicity of prepared compounds could also facilitate passage through the mycobacterial cell wall.
Acknowledgements
This work was financially supported by GAUK B-CH/710312, and IGA NT 13346. The publication is a result of the project implementation: Support of establishment, development, and mobility of quality research teams at the Charles University, Project Number CZ.1.07/2.3.00/30.20.0235, supported by The Education for Competitiveness Operational Programme (ECOP) and co-financed by the European Social Fund and the state budget of the Czech Republic. Authors wish to thank Ida Dufková for performing in vitro antifungal and antibacterial screening, and Assoc. Prof. Jiří Kuneš for recording of NMR spectra.
Conflicts of interest: none.
Received 14 Februar 2013 / Accepted 5 March 2013
Prof. PharmDr. Martin Doležal, Ph.D., J. Vobicková, B. Servusová
Charles University in Prague, Faculty of Pharmacy,
Department of Pharmaceutical Chemistry and Drug Control
Heyrovského 1203, 500 05 Hradec Králové, Czech Republic
e-mail: martin.dolezal@faf.cuni.cz
P. Paterová
Department of Clinical Microbiology, University Hospital, Hradec Králové, Czech Republic
Sources
1. WHO. Global Tuberculosis Report 2012. Available online: http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf (accessed on 14th February 2013).
2. Dalmer O., Eugen W. (E. Merck) Pyrazine Derivatives. 1936, DE 632257.
3. Hall S. A., Spoerri P. E. Syntheses in the Pyrazine Series. II. Preparation and Properties of Aminopyrazine. J Am Chem Soc 1940; 62, 664–665.
4. Dessau F. I., Burger F. J., Yeager R. L., Kulish M. A Method for the Determination of in vitro Sensitivity of Tubercle Bacilli to Pyrazinamide (Aldinamide). Am Rev Tuberc 1952; 65, 635–636.
5. Yeager R. L., Munroe W. G. C., Dessau F. I. Pyrazinamide (Aldinamide) in the Treatment of Pulmonary Tuberculosis. Am Rev Tuberc 1952; 65, 523–545.
6. Malone L., Schurr A., Lindh H., McKenzie D., Kiser J. S., Williams J. H. The Effect of Pyrazinamide (Aldinamide) on Experimental Tuberculosis in Mice. Am Rev Tuberc 1952; 65, 511–518.
7. Solotorovsky M., Gregory F. J., Ironson E. J., Bugie E. J., O’Neill R. C., Pfister K. Pyrazinoic Acid Amide; an Agent Active against Experimental Murine Tuberculosis. Proc Soc Exp Biol Med 1952; 79, 563–565.
8. Kushner S., Dalalian H., Sanjurjo J. L., Bach F. L. Jr., Safir S. R., Smith V. K. Jr., Williams J. H. Experimental Chemotherapy of Tuberculosis. 2. The Synthesis of Pyrazinamides and Related Compounds. J Am Chem Soc 1952; 74, 3617–3621.
9. Konno K., Feldmann F. M., McDermott, W. Pyrazinamide Susceptibility and Amidase Activity of Tubercle Bacilli. Am Rev Respir Dis 1967; 95, 461–469.
10. Zhang Y., Mitchison D. The Curious Characteristics of Pyrazinamide: a Review. Int J Tuberc Lung Dis 2003; 7, 6–21.
11. Mitchison D. A. The Action of Antituberculosis Drugs in Short Course Chemotherapy. Tubercle 1985; 66, 219–225.
12. Ibrahim M., Andries K., Lounis N., Chauffour A., Truffot-Pernot C., Jarlier V., Veziris N. Synergistic Activity of R207910 Combined with Pyrazinamide against Murine Tuberculosis. Antimicrob Agents Chemother 2007; 51, 1011–1015.
13. Diacon A. H., Pym A., Grobusch M., Patientia R., Rustomjee R., Page-Shipp L., Pistorius Ch., Krause R., Bogoshi M., Churchyard G., Venter A., Allen J., Palomino J. C., De Marez T., van Heeswijk R. P. G., Lounis N., Meyvisch P., Verbeeck J., Parys W., de Beule K., Andries K., Mc Neeley D. F. The Diarylquinoline TMC207 for Multidrug-Resistant Tuberculosis. N Engl J Med 2009; 360 : 2397–2405.
14. Doležal M., Kešetovič D., Zitko J. Antimycobacterial Evaluation of Pyrazinecarboxylic Acid Derivatives. Curr Pharm Design 2011; 17, 3506–3514.
15. Cynamon M. H., Klemens S. P., Chou T. S., Gimi R. H., Welch J. T. Antimycobacterial Activity of a Series of Pyrazinoic Acid-Esters. J Med Chem 1992; 35, 1212–1215.
16. Cynamon M. H., Gimi R., Gyenes F., Sharpe C. A., Bergmann K. E., Han H. J., Gregor L. B., Rapolu R., Luciano G., Welch J. T. Pyrazinecarboxylate Esters with Broad Spectrum in vitro Antimycobacterial Activity. J Med Chem 1995; 38, 3902–3907.
17. Cynamon M. H., Welch J. T. Preparation of Pyrazinoic Acid Esters as Anti-Mycobacterium avium Agents. U.S. Pat. 1997, US 5643912 A 19970701.
18. Bergmann K. E., Cynamon M. H., Welch J. T. Quantitative Structure-Activity Relationships for the in vitro Antimycobacterial Activity of Pyrazinoic Acid Esters. J Med Chem 1996; 39, 3394–3400.
19. Fernandes J. P. S., Pasqualoto K. F. M., Felli V. M. A., Ferreira E. I., Brandt C. A. QSAR Modeling of a Set of Pyrazinoate Esters as Antituberculosis Prodrugs. Arch Pharm (Weinheim, Germany) 2010; 343, 91–97.
20. Yamamoto S., Toida I., Watanabe N., Ura T. In vitro Antimycobacterial Activities of Pyrazinamide Analogs. Results of Screening Tests. Kekkaku 1996; 71, 253–258.
21. Seitz L. E., Suling W. J., Reynolds R. C. Synthesis and Antimycobacterial Activity of Pyrazine and Quinoxaline Derivatives. J Med Chem 2002; 45, 5604–5606.
22. Terasawa T., Shigenaga S., Itoh S., Maeda J., Watanabe H., Kubo S., Ishii N. Preparation of Heterocyclic Carboxamide Compounds, in Particular Nicotinamides as ROCK Inhibitors. PCT Int. Appl. 2010, WO 2010032875 A2 20100325.
23. Chen S., Corbett W. L., Guertin K. R., Haynes N. E., Kester R. F., Mennona F. A., Mischke S. G., Qian Y., Sarabu R., Scott N. R., Thakkar K. C. Preparation of Pyrazines and Related Compounds as Glucokinase Activators for the Treatment of Type II Diabetes. PCT Int. Appl. (2004), WO 2004052869 A1 20040624.
24. Caudill J., Cooney M., Nigam S. C. Preparation of 5-Methylpyrazine-2-carboxylic Acid 4-Oxide Esters and Salts via Oxidation Using Oxone in a Halogenated Solvent. U.S. Pat. Appl. Publ. 2005, US 20050239803 A1 20051027.
25. Sayahi H., Pugliese K. M., Zimhony O., Jacobs W. R. Jr., Shekhtman A., Welch J. T. Analogs of the Antituberculous Agent Pyrazinamide Are Competitive Inhibitors of NADPH Binding to M. tuberculosis Fatty Acid Synthase I. Chem Biodivers 2012; 9, 2582–2596.
26. Ngo S. C., Zimhony O., Chung W. J., Sayahi H., Jacobs W. R. Jr., Welch J. T. Inhibition of Isolated Mycobacterium tuberculosis Fatty Acid Synthase I by Pyrazinamide Analogs. Antimicrob Agents Chemother 2007; 51, 2430–2435.
27. Weijlard J., Tishler M., Erickson A. E. New Aminopyrazines and their Sulfanilamide Derivatives. J Am Chem Soc 1945; 67, 802–806.
28. Vontor T., Palát K., Lyčka A. Homolytic Carbamoylation and Alkoxycarbonylation of 2-Aminopyrazine. Coll Czech Chem Commun 1989; 54, 1306–1310.
29. Simões M. F., Valente E., Gómez J. R. M., Anes E., Constantino L. Lipophilic Pyrazinoic Acid Amide and Ester Prodrugs Stability, Activation and Activity against M. tuberculosis. Eur J Pharm Sci 2009; 37, 257–263.
30. Jones R. N., Barry A. L. Optimal Dilution Susceptibility Testing Conditions, Recommendations for MIC Interpretation, and Quality-Control Guidelines for the Ampicillin-Sulbactam Combination. J Clin Microbiol 1987; 25, 1920–1925.
31. Zitko J., Doležal M., Svobodová M., Vejsová M., Kuneš J., Kučera R., Jílek P. Synthesis and Antimycobacterial Properties of N-Substituted 6-Amino-5-cyanopyrazine-2-carboxamides. Bioorg Med Chem 2011; 19, 1471–1476.
32. Servusová B., Eibinová D., Doležal M., Kubíček V., Paterová P., Peško M., Kráľová K. Substituted N-Benzylpyrazine-2-carboxamides: Synthesis and Biological Evaluation. Molecules 2012; 17, 13183–13198.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2013 Issue 2
Most read in this issue
- Evaluation of the influence of sterilization method on the stability of carboxymethyl cellulose wound dressing
- Prolegomenon of the Czech pharmacognosy: 21st century
-
Physiological aspects of lipoxygenase in plant signaling systems
Part I. Octadecanoid pathway - Influence of the degree of substitution on the absorptivity of acidic carboxymethyl cellulose in the form of nonwoven fabric