
Účinná imunoterapie glioblastomu u adolescenta se syndromem konstitučního deficitu v mismatch repair opravném systému
Authors:
Z. Pavelka 1; K. Zitterbart 1; H. Nosková 1,2; V. Bajčiová 1; O. Slabý 2; J. Štěrba 1
Authors‘ workplace:
Klinika dětské onkologie LF MU a FN Brno
1; CEITEC – Středoevropský technologický institut, Masarykova univerzita, Brno
2
Published in:
Klin Onkol 2019; 32(1): 70-74
Category:
Case Report
doi:
https://doi.org/10.14735/amko201970
Overview
Východiska:
Jedinci s konstitučním deficitem v mismatch repair opravném systému (constitutional mismatch repair-deficiency syndrome – CMMR-D) jsou charakterizováni časným výskytem karcinomu tlustého střeva, hematologických malignit a nádorů mozku (maligní gliomy, gliomy vysokého stupně malignity) v dětství, adolescenci či rané dospělosti. Vysoká mutační nálož v nádoru, typická pro glioblastom u pacientů s CMMR-D, může být příčinou pozitivní léčebné odpovědi na imunoterapii vč. checkpoint inhibitorů.
Případ:
Podáváme kazuistiku adolescenta s prokázaným CMMR-D metodou celoexomového sekvenování (mutace c.2T>A/ p.M1K a c.2521delT/p.W841fs genu PMS2), který postupně onemocněl karcinomem tlustého střeva a glioblastomem s vysokou mutační náloží. Součástí individualizované terapie glioblastomu na základě biologického profilu nádoru byla kromě radioterapie s metronomickým temozolomidem i imunoterapie – vakcinace autologní vakcínou z dendritických buněk produkujících IL-12 a nivolumab. Pacient žije 21 měsíců od diagnózy glioblastomu s kompletní léčebnou odpovědí dokumentovanou na opakovaných vyšetřeních magnetickou rezonancí.
Závěr:
Jedince s CMMR-D je třeba pravidelně sledovat dle zavedených algoritmů. Mezi nimi hraje zásadní roli celotělová magnetická rezonance, která umožňuje záchyt malignity v časném období. Nádorová onemocnění u pacientů s CMMR-D vykazují zpravidla hypermutovaný genotyp a mohou odpovídat na imunoterapii. Konvenční léčba glioblastomu je pouze paliativní, nemocní mohou profitovat z individuálního léčebného přístupu na základě znalosti biologického profilu nádoru. Podmínkou je extenzivní vyšetření nádorové tkáně metodami molekulární biologie.
Klíčová slova
vrozené nádorové predispoziční syndromy – glioblastom – celoexomové sekvenování – imunoterapie – vakcíny – inhibitory kontrolních bodů
Úvod
Podkladem autozomálně recesivního syndromu konstitučního deficitu v mismatch repair opravném systému (constitutional mismatch repair-deficiency syndrome – CMMR-D) jsou bialelické zárodečné mutace nejčastěji v genech PMS2 a MSH6 [1–4]. Nositelé CMMR-D jsou charakterizováni nálezem skvrn café-au-lait (café-au-lait macules – CALM), hypopigmentací, střevními polypy a výskytem malignit typického spektra v dětství, adolescenci a časné dospělosti. Nejčastější jsou nádory mozku (maligní gliomy) zpravidla v první dekádě, gastrointestinální tumory (většinou kolorektální karcinom) v druhé dekádě a následují hematologické malignity (lymfomy, leukemie). Častý je výskyt vícero typů malignit u téhož jedince, přičemž nádory mohou být synchronní nebo metachronní [1–5]. Nositelé CMMR-D mají být pravidelně sledováni podle vypracovaných algoritmů; specifickou roli hraje celotělové vyšetření magnetickou rezonancí (whole body magnetic resonance imaging – WBMRI), které umožňuje časný záchyt nádoru [5].
Nedávno bylo zjištěno, že nádory v rámci CMMR-D vykazují univerzálně vysokou mutační nálož (> 100 mutací/ Mb) ve srovnání s běžnými sporadickými nádory [6]. Zvýšená genomická instabilita může být podkladem pro odlišnou efektivitu konvenční genotoxické terapie, a naopak pro léčebné odpovědi na imunoterapii. CMMR-D postihuje primárně replikaci DNA a nejsou popsány případy excesivní toxicity chemoterapie nebo radioterapie [6].
Popis případu
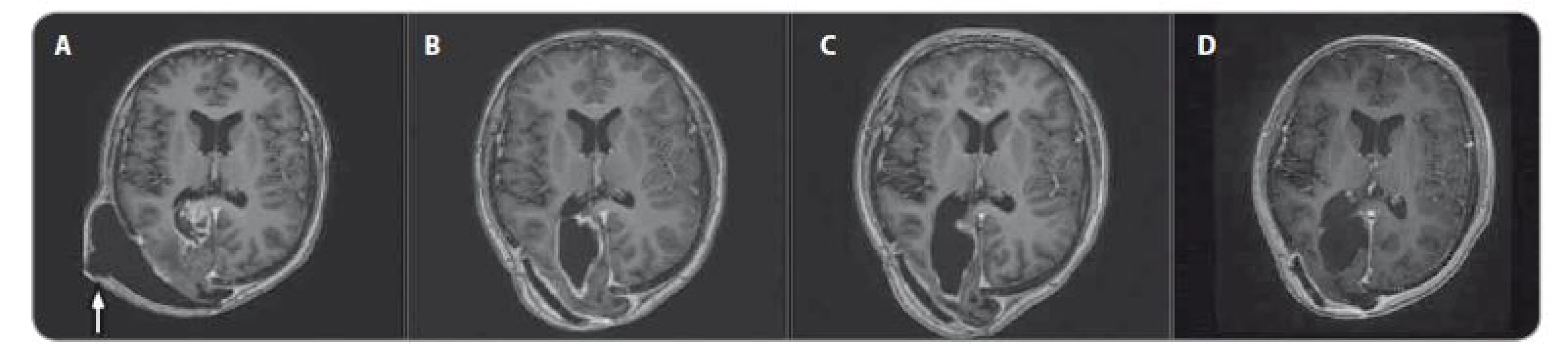
U chlapce ve věku 14 let byl diagnostikován bifokální invazivní adenokarcinom kolon (G2, pT2, pN1a, pM0, klinické stadium IIIA) s mutací KRAS, léčen byl hemikolektomií a dvanácti cykly FOLFOX. Léčba navodila trvající kompletní remisi. Výskyt „adultního“ typu nádoru u adolescenta vedl ke genetickému vyšetření. Metodou celoexomového sekvenování (whole exome sequencing – WES) byla u pacienta prokázána kombinace zárodečných mutací c.2T>A/ p.M1K a c.2521delT/p.W841fs genu PMS2 v heterozygotním stavu, a tím potvrzen bialelický CMMR-D s autozomálně recesivním typem dědičnosti (obr. 1, 2). Rodinná studie nalezla heterozygotní mutaci c.2T>A/ p.M1K u otce probanda, heterozygotní mutaci c.2521delT/p.W841fs genu PMS2 u matky probanda a heterozygotní mutace c.2T>A/ p.M1K a c.2521delT/p.W841fs genu PMS2 u bratra probanda (ročník 2007). Nejedná se tedy o nové, ale zděděné mutace vždy po jedné od každého z rodičů. Oba rodiče mají negativní onkologickou anamnézu. Klinicky je u pacienta nalezena 1 CALM, polypy tlustého střeva, další kožní projevy CMMR-D nezachyceny. Pacient byl sledován vč. opakovaných tumor markerů, endoskopií gastrointestinálního traktu, ultrazvuku břicha a střev a celotělových MRI vyšetření. V únoru 2017 nejprve celotělové MRI a následné cílené MRI mozku (obr. 3) zachytilo asymptomatický supratentoriální tumor vpravo. Neurochirurg provedl subtotální resekci s ponecháním drobného makroskopického rezidua v corpus callosum, histopatologický nález potvrdil glioblastom s přítomností extenzivní primitivní neuronální komponenty. Tkáň tumoru byla vyšetřena metodou WES, analýza somatického exomu tumoru prokázála velmi vysokou mutační nálož (265 mutací/ Mb), resp. hypermutovaný stav (vč. mutací PIK3CA, PIK3R1, PDGFRA, PDGFRB, KDR, BRCA2, DNMT3A, TP53, RET, PTEN). Z tkáně tumoru byl připraven lyzát k výrobě autologní dendritické vakcíny, následně proběhla leukaferéza a po příslušné laboratorní fázi výroby byla k dispozici autologní vakcína dendritických buněk (dendritic cells – DC) produkujících IL-12 (vše v rámci řádné akademické studie fáze I KDO_DC1311 LF MU). Adjuvantní terapie – nejprve lokální radioterapie (59,4 Gy) + konkomitantní chemoterapie s temozolomidem (TMZ) 75 mg/ m2/ den (Stuppův režim). V dalším období se znalostí analýzy WES postup na základě individuálního biologického profilu tumoru – imunoterapie – vakcinace autologní DC vakcínou (celkem 5× každých 14 dní) + nivolumab 3 mg/ kg intravenózně každých 14 dní + TMZ v metronomickém dávkování 20 mg/ m2/ den (21 dní „on“, 7 dní „off“ opakovaně, nikoliv bloková terapie). Nivolumab je podáván v rámci „off label“ indikace. Tolerance terapie je dobrá, jedná se o ambulantní režim s vysokou kvalitou života, dosud se nevyskytla žádná toxicita stupně 3 nebo 4, žádná imunologická toxicita. Kontrolní MRI mozku prokazují pokračující regresi, po 15 měsících terapie již kompletní, přežití bez příhody 21 měsíců (obr. 4).



Diskuze
Jedince s CMMR-D je třeba sledovat dle zavedených algoritmů, mezi nimi má výsadní postavení opakované WBMRI, které umožňuje záchyt malignity v časném období [5]. Sporadický glioblastoma multiforme (GBM) je onemocnění konvenční terapií zpravidla neléčitelné [7]. Klasický Stuppův režim (radioterapie + TMZ) je paliativní [8], přidání bevacizumabu k této standardní terapii dospělých nepřineslo pro dětské pacienty žádnou výhodu [9]. Na našem pracovišti léčíme děti či adolescenty s prognosticky nepříznivou diagnózou (dlouhodobé přežívání pod 30 %), mezi které glioblastom patří, již v první linii individuálně na základě biologického profilu nádoru v rámci tzv. personalizované medicíny [10]. Máme za to, že konvenční onkologická terapie je u takových diagnóz (gliomy mozkového kmene, glioblastom, atypický teratoidní/ rhabdoidní tumor, metastatické sarkomy, některé metastatické neuroblastomy) neefektivní, či pouze paliativní. Konkrétní terapeutické doporučení je formulováno panelem odborníků (klinický onkolog, molekulární biolog, klinický genetik, farmakolog) na tzv. molekulárně onkologické komisi konané zpravidla jednou měsíčně. Na podkladě vyšetření nádorové tkáně zejména metodou celoexomového sekvenování doplněného o vyšetření genových expresí získáme individuální profil mutací či alterací jednotlivých signálních drah nádoru pacienta, data jsou porovnána s databází a výsledkem je návrh potenciálních léčebných cílů a jejich inhibitorů [11]. Cílená biologika jsou po zhodnocení interakcí a možné toxicity nasazena buď formou augmentace konvenční léčby, nebo v udržovací fázi [10,11]. Výsledky molekulárních vyšetření jsou k dispozici s jistou latencí, proto prvním krokem léčby byla konvenční chemoradioterapie a individuální postup byl uplatněn v adjuvantní fázi. Současný koncept tvrdí, že vysoká mutační nálož nádorů pacientů s CMMR-D je podkladem pro léčebné odpovědi při imunoterapii checkpoint inhibitory díky vysokému stupni prezentace neoantigenů aktivujících T lymfocyty [6]. Dalším směrem imunoterapie, která prokázala u selektované podskupiny pacientů s gliomy vysokého stupně malignity (high-grade glioma – HGG) a minimálním reziduem prodloužení přežívání, je protinádorová vakcinace na bázi autologních dendritických buněk [6,12–14]. Akasaki et al prezentovali prodloužené přežívání pacientů s nově diagnostikovaným GBM léčených kombinací TMZ a vakcinace dendritickými buňkami – přežití bez progrese 18,3 měsíce, celkové přežití 30,5 měsíce [15]. Naše pracoviště realizuje studii fáze I „Kombinovaná protinádorová terapie s autologní vakcínou z dendritických buněk produkujících interleukin 12 u dětských a adolescentních pacientů s progredujícími, relabujícími nebo primárně metastatickými malignitami vysokého rizika KDO_DC1311“ (EudraCT number 2014-003388-39). U našeho pacienta na základě biologické analýzy tumoru (k dispozici jsou detailní výsledky transkriptomu, somatického i germinálního exomu, genových fúzí, fosforylace kináz, mutační nálož) byla molekulární onkologickou komisí doporučena terapie kombinací metronomicky podávaného TMZ s imunoterapií (nivolumab + vakcinace autologní dendritickou vakcínou). Metronomické dávkování TMZ umožňuje vyhnout se významné leukopenii/ lymfopenii, která by interferovala s mechanizmem účinku imunoterapie [6,16]. Dosažení pozitivní léčebné odpovědi koreluje s publikovanými daty, která podporují lepší efektivitu imunoterapeutických postupů u pacientů s hypermutovanými malignitami v rámci CMMR-D proti konvenční terapii [6,13–15]. Tito pacienti také mají publikováno delší průměrné přežívání (v jedné práci > 27 měsíců) v porovnání se sporadickými případy GBM (12 měsíců), jednalo se zejména o pacienty s bialelickými zárodečnými mutacemi PMS2 genu [4,6,13–15].
Závěr
Tato kazuistika podporuje úvahu, že pacienti s glioblastomem v rámci CMMR-D díky hypermutovanému genotypu mohou profitovat z imunoterapie lépe než z konvenční léčby. V současnosti je na Klinice dětské onkologie LF MU a FN Brno připravována akademická studie testující kombinovanou terapii autologní dendritickou vakcínou produkující IL-12 a nivolumabem u vybraných pediatrických malignit vysokého rizika vč. glioblastomu.
Práce byla podpořena grantovým projektem Ministerstva zdravotnictví ČR AZV 16-33209A (Sekvenování nové generace a expresní profilovaní jako diagnostický podklad pro návrhy individualizovaných léčebných plánů pro děti se solidními nádory).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Zdeněk Pavelka
Klinika dětské onkologie LF MU a FN Brno
Černopolní 9
613 00 Brno
e-mail: pavelka.zdenek@fnbrno.cz
Obdrženo: 26. 9. 2018
Přijato: 18. 11. 2018
Sources
1. Strahm B, Malkin D. Hereditary cancer predisposition in children: genetic basis and clinical implications. Int J Cancer 2006; 119(9): 2001–2006. doi: 10.1002/ ijc.21962.
2. Wimmer K, Kratz CP. Constitutional mismacth repair-deficiency syndrome. Haematologica 2010; 95(5): 699–701. doi: 10.3324/ haematol.2009.021626.
3. Cavenee WK, Hawkins C, Burger PC et al. Turcot syndrome. In: Louis DN, Ohgaki H, Wiestler OD et al (eds). WHO classification of tumors of the central nervous system. Lyon: International Agency for Research on Cancer 2016 : 317–318.
4. De Vos M, Hayward BE, Charlton R et al. PMS2 mutation in childhood cancer. J Natl Cancer Inst 2006; 98(5): 358–361. doi: 10.1093/ jnci/ djj073.
5. Tabori U, Hansford JR, Achatz MI et al. Clinical management and tumor surveillance recommendations of inherited mismacth repair deficiency in childhood. Clin Cancer Res 2017; 23(11): e32–e37. doi: 10.1158/ 1078-0432.CCR-17-0574.
6. Bouffet E, Larouche V, Campbell BB et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 2016; 34(19): 2206–2211. doi: 10.1200/ JCO.2016.66.6552.
7. Vanan MI, Mehta V, Eisenstat DD. High-Grade Glioma. In: Scheinemann K, Bouffet E (eds). Pediatric neuro-oncology. New York: Springer 2015 : 101–116.
8. Stupp R, Mason WP, van den Bent MJ et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005; 352(10): 987–996. doi: 10.1056/ NEJMoa043330.
9. Grill J, Massimino L, Bouffet E et al. Phase II, open-label, randomized, multicenter trial (HERBY) of bevacizumab in pediatric patients with newly diagnosed high-grade glioma. J Clin Oncol 2018; 36(10): 951–958. doi: 10.1200/ JCO.2017.76.0611.
10. Pearson AD, Herold R, Rousseau R et al. Implementation of mechanism of action biology-driven early drug development for children with cancer. Eur J Cancer 2016; 62 : 124–131. doi: 10.1016/ j.ejca.2016.04.001.
11. Hyman DM, Solit DB, Arcila ME et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today 2015; 20(12): 1422–1428. doi: 10.1016/ j.drudis.2015.08.005.
12. Bajčiová V. Role imunoterapie v dětské onkologii. Klin Onkol 2015; 28 (Suppl 4): 4S38–4S43. doi: 10.14735/ amko20154S38.
13. Weiss T, Weller M, Roth P. Immunotherapy for glioblastoma: concepts and challenges. Curr Opin Neurol 2015; 28(6): 639–646. doi: 10.1097/ WCO.0000000000000249.
14. Johanns TM, Miller CA, Dorward IG et al. Immunogenomics of hypermutated glioblastoma: a patient with germline POLE deficiency treated with checkpoint blockade immunotherapy. Cancer Discov 2016; 6(11): 1230–1236. doi: 10.1158/ 2159-8290.CD-16-0575.
15. Akasaki Y, Kikuchi T, Homma S et al. Phase I/ II trial of combination of temozolomide chemotherapy and immunotherapy with fusions of dendritic and glioma cells in patients with glioblastoma. Cancer Immunol Immunother 2016; 65(12): 1499–1509. doi: 10.1007/ s00262-016-1905-7.
16. Sterba J, Pavelka Z, Slampa P. Concomitant radiotherapy and metronomic temozolomide in pediatric high-risk brain tumors. Neoplasma 2002; 49(2): 117–1120.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2019 Issue 1
Most read in this issue
- Zhubné nádory penisu – diagnostika a liečba
- Extrakraniální stereotaktická radioterapie – přehled současných indikací
- Vliv velikosti nádorové masy a stavu p16 na léčebné výsledky – dosažení kompletní remise u prospektivně sledovaných pacientů s nádory orofaryngu
- Mají subtypy HER2 pozitivního karcinomu prsu význam pro klinickou praxi?