
Aplikace molekulární diagnostiky kardiomyopatií v pediatrické praxi
Authors:
L. Skutková 1; D. Humlová 1,2; J. Kadlecová 3
Authors‘ workplace:
Pediatrická klinika LF MU a FN Brno
1; Centrum kardiovaskulární a transplantační chirurgie, Brno
2; Cytogenetická laboratoř Brno, s. r. o.
3
Published in:
Kardiol Rev Int Med 2016, 18(4): 268-271
Overview
Dědičné typy kardiomyopatií patří k nejčastějším dědičným vadám kardiovaskulárního systému. V posledních letech se genetické vyšetření stává součástí diagnostiky těchto onemocnění. Je známo více než 80 genů, které jsou se vznikem kardiomyopatií asociovány, a dle nejnovějších poznatků se jejich patofyziologický podklad překrývá s podstatou kanalopatií. Pomocí moderních molekulárně-genetických metod se rozšířilo spektrum vyšetřovaných genů a v naší laboratoři je v současné době vyšetřováno zároveň 97 genů zavzatých do patogeneze těchto onemocnění. Na základě molekulárně genetické diagnostiky je možné upřesnit diagnózu u pacientů s kardiomyopatií a především identifikovat presymptomatické jedince v rodině. Děti, které zdědily suspektní variantu, jsou pravidelně kardiologicky sledovány stejně tak jako příbuzní probandů, u kterých nebyla genetická podstata onemocnění objasněna.
Klíčová slova:
kardiomyopatie – molekulární diagnostika – genetika – pediatrie
Úvod
Kardiomyopatie jsou charakterizované poruchou funkce nebo struktury srdečního svalu. Jedná se o heterogenní skupinu onemocnění s různými klinickými příznaky, které se často vyvíjí pomalu a v konečném stadiu mohou vést k srdečnímu selhání (SS). Jsou také známým rizikovým faktorem pro náhlou srdeční smrt, typicky u mladých sportovců. Onemocněním jsou častěji postiženi dospělí, může se však projevit v jakémkoli věku. V pediatrické populaci je maximum výskytu ve skupině dětí do jednoho roku věku a poté v pubertě.
Podle etiologie lze kardiomyopatie rozdělit na primární a sekundární. U sekundárních typů je postižení srdce důsledkem jiného onemocnění nebo působením toxických látek. V dětském věku jsou to často kardiomyopatie spojené s infekcí, vrozenými srdečními vadami, chemoterapií, svalovými dystrofiemi nebo endokrinními poruchami. Vyskytují se také u vzácných metabolických poruch, např. u některých střádavých onemocnění a mitochondriálních vad, nebo jako součást genetických syndromů, např. Noonanův, Carvajalův či Barthův syndrom.
U primárních forem je příčina nejasná a často se jedná o dědičná onemocnění. V tomto článku budou popsány současné možnosti genetického vyšetření dědičných typů kardiomyopatií a na vybrané kazuistice bude ukázán příklad interpretace výsledků molekulárně-genetické analýzy.
Genetika dědičných kardiomyopatií
Dědičné typy kardiomyopatií patří k nejčastějším dědičným vadám kardiovaskulárního (KV) systému. Jejich výskyt je často familiární, kdy se onemocnění projevuje v několika generacích postižené rodiny, existují však i sporadické formy s de novo mutací u probanda. Familiární kardiomyopatie jsou většinou monogenní, autozomálně dominantně dědičná onemocnění. Jsou ale známy i formy autozomálně recesivní, X-vázané a typy s maternální dědičností [1].
Tato skupina onemocnění je známá vysokou fenotypovou a genetickou variabilitou. To znamená, že stejný fenotyp může být podmíněn velkým počtem různých mutací a na druhou stranu mutace ve stejném genu mohou vést k různým projevům onemocnění srdečního svalu i k různým typům kardiomyopatií. Stejná genová mutace u jednoho pacienta může být maligní s vysokým rizikem náhlé srdeční smrti, u jiného pacienta se nemusí projevit vůbec. To ukazuje na multifaktoriální příčinu vzniku tohoto onemocnění, vč. vlivu prostředí [2]. Svou roli na penetranci hraje i věk nemocného. Konkrétní kauzální mutace jsou v populaci většinou velmi vzácné a jsou privátní pro danou rodinu. Proto není užitečný screening známých mutací, ale je nutné se zaměřit na celé geny.
V poslední době se ukazuje, že genetický a patofyziologický podklad dědičných kardiomyopatií se ve značné míře překrývá s podstatou dědičných forem srdečních arytmií – kanalopatií, jako např. syndrom short QT, syndrom long QT, Brugada syndrom, syndrom časné repolarizace, katecholaminergní polymorfní ventrikulární tachykardie a některé případy syndromu náhlého úmrtí kojence [3,4].
Současné možnosti molekulární diagnostiky kardiomyopatií
S dědičným typem kardiomyopatií je asociováno více než 80 různých genů s vysokou variabilitou a různou penetrancí. Vzhledem k této značné genetické heterogenitě byla do nedávné doby diagnostika kardiomyopatií na molekulární úrovni velice omezená. S příchodem technologie masivního paralelního sekvenování se otevřela možnost analyzovat současně velký počet genů za krátkou dobu a ve srovnání s klasickým sekvenováním za relativně nízkou cenu a s dobrým diagnostickým přínosem.
Od počátku roku 2016 byla molekulárně-genetická diagnostika kardiomyopatií a kanalopatií v Cytogenetické laboratoři Brno, s. r. o., sloučena a u pacientů s těmito diagnózami je analyzováno 97 vybraných genů, které jsou v souvislosti s rozvojem těchto onemocnění nejčastěji popisovány. K vyšetření jsou indikováni pacienti s prokázanou kardiomyopatií nebo s pozitivní rodinnou anamnézou. Všichni podstoupí vyšetření klinickým genetikem a je sestaven rodokmen probanda. Po poučení o vyšetření a po podepsání informovaného souhlasu ke genetickému testování je proveden odběr periferní krve a izolace DNA z lymfocytů k molekulárně-genetickému vyšetření. Následně je cíleně sekvenováno 97 genů zapojených do patogeneze kardiomyopatií a kanalopatií metodou sekvenování nové generace (next generation sequencing – NGS). Klinický význam nalezených variant je hodnocen na základě jejich četnosti v populaci, vlivu varianty na výsledný protein, výstupů z predikčních programů a informací z mezinárodních databází a vědeckých publikací. Podle klinického významu jsou nalezené varianty DNA rozděleny na základě směrnic The American College of Medical Genetics and Genomics (ACMG) do pěti tříd: patogenní, potenciálně patogenní, varianty nejasného významu (variant of unknown significance – VOUS), potenciálně benigní a benigní [5].
Suspektní mutace se ověřují klasickým Sangerovým sekvenováním z nezávislého odběru vzorku z bukálního stěru. V případě zjištění pravděpodobné kauzální mutace u probanda se provádí segregace mutace v rodině.
Kazuistika chlapce s familiární formou dilatační kardiomyopatie
Tato kazuistika pojednává o 15letém chlapci s pozitivní rodinnou anamnézou na dilatační kardiomyopatii (DCM). Jeho matka má projevy DCM od 31 let, matka matky zemřela v důsledku DCM ve 29 letech, bratr matky zemřel na DCM v 33 letech, sestra matky zemřela na DCM ve 42 letech, její syn podstoupil v 33 letech transplantaci srdce. Pacient má dva bratry, starší je po transplantaci srdce ve 14 letech, druhý byl na vyšetření ve 20 letech asymptomatický. Otec a příbuzenstvo z jeho strany jsou bez srdečních potíží. Chlapec chodil do deváté třídy základní školy, měl průměrný prospěch, hrál florbal a stolní tenis. Byl pravidelně preventivně sledován na kardiologii. Ve 14 letech byl zjištěn pokles ejekční frakce (EF) o 20 % a současně byly přítomny známky dekompenzovaného pravostranného srdečního selhávání (dyspepsie, malý pleurální a perikardiální výpotek). Byla zahájena léčba chronického SS. Jeho stav rychle progredoval, za dva měsíce byla EF levé komory 20 %, na ultrazvuku srdce byla patrná těžká systolická i diastolická dysfunkce levé i pravé komory, středně významná mitrální regurgitace, významná trikuspidální regurgitace a plicní hypertenze. Pacient byl zařazen na urgentní waiting list k transplantaci srdce. Krátce nato mu byl z důvodu běhu nesetrvalých komorových tachykardií implantován kardioverter-defibrilátor (ICD). Postupně byla nutná intermitentní a poté i kontinuální intravenózní podpora inotropiky a diuretiky. Za pět měsíců od prvních klinických příznaků DCM podstoupil chlapec transplantaci srdce.
Vzhledem k pozitivní rodinné anamnéze a jisté familiární formě DCM bylo indikováno genetické vyšetření, sestaven rodokmen (obr. 1) a byla provedena mutační analýza 46 genů asociovaných s kardiomyopatií pomocí NGS. Byla aplikována metodika s využitím kytu TruSightCardiomyopathy. V analyzovaném vzorku bylo detekováno celkem 68 variant, které vyhovovaly filtru pokrytí (coverage) minimálně 20× a frekvenci varianty min. 20 %. Podle předpokládaného klinického významu byly varianty rozděleny na jednu potenciálně patogenní variantu, devět variant nejasného významu, dvě potenciálně benigní a 56 benigních variant.
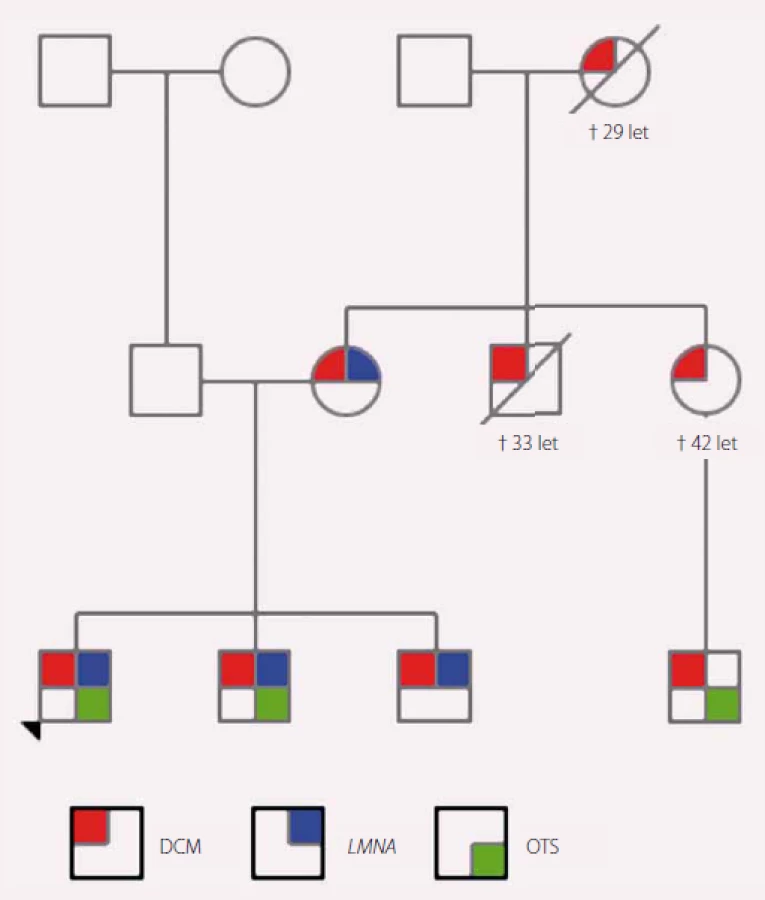
Jako potenciálně patogenní byla klasifikována varianta v genu LMNA, (chromozom 1): NM_170707.3:c.1621C>T, NP_733821.1:p.Arg541Cys, genotyp c.[1621C>T]+[=], typ mutace missense. Podle výsledku predikčních programů SIFT a PolyPhen je tato varianta v heterozygotním stavu klasifikována jako potenciálně patogenní (deleterious(0):SIFT/ probably damaging(1):PolyPhen). V literatuře již byla tato změna popsána jako varianta asociovaná se segmentální akinezou/ dyskinezou svaloviny levé komory [6] a elektrokardiografickými abnormalitami (neurčená intraventrikulární blokáda, patologický Q kmit, ventrikulární arytmie) [7]. Patogenita byla potvrzena transfekcí u experimentálních myoblastů [7].
Výsledek z NGS u našeho pacienta byl verifikován Sangerovým sekvenováním (obr. 2) a bylo doporučeno testování i přímých příbuzných. Postupně byli vyšetřeni oba bratři a matka. U všech byla nalezena stejná sekvenční změna. Mladší bratr byl v době vyšetření probanda bez echokardiografických známek DCM, na kontrole za 12 měsíců již byla zjištěna snížená EF levé komory. Vzhledem k rodinné anamnéze, pozitivitě molekulárně-genetického vyšetření a dostupných informací o této variantě lze předpokládat progresivní průběh DCM i u tohoto pacienta.

Tato kazuistika je příkladem rodiny, kde byla nalezena mutace, která již byla v databázích popsána v asociaci s onemocněním. Pomocí sestavení rodokmenu a následné segregační analýzy pokrevních příbuzných byla její kauzalita v tomto případě potvrzena.
V současné době probíhá vyšetření dalších přímých příbuzných k detekci presymptomatických jedinců nebo naopak k vyloučení nosičství patogenní varianty.
Závěr
V posledních letech vzrůstá význam molekulárně-genetického vyšetření u nemocných trpících různými typy kardiomyopatií a jejich rodin. Toto vyšetření upřesňuje diagnózu a v některých případech i prognózu a pomáhá určit riziko výskytu onemocnění u příbuzných. S přihlédnutím k převážně autozomálně dominantnímu typu dědičnosti, tedy 50% riziku přenosu na potomky, by měli být vyšetřeni všichni příbuzní probanda z první, druhé a třetí generace. U všech by mělo být provedeno kardiologické vyšetření zahrnující fyzikální vyšetření, echokardiografii, EKG, v indikovaných případech další pomocné metody jako MR, ergometrie, Holterovo monitorování a podobně. Příbuzní by také měli podstoupit genetické poradenství. Pokud je odhalena mutace nebo více variant odpovědných za dané onemocnění, je možné provést molekulární diagnostiku i u příbuzných a zahájit včasnou profylaktickou terapii. Při negativním výsledku u příbuzného lze naopak pacienta vyřadit ze sledování.
Analýza 97 genů zapojených do patogeneze kardiomyopatií a kanalopatií metodou NGS pomohla rozšířit možnosti molekulárně genetického testování. Přesto se u některých pacientů nepodaří nalézt patologickou variantu asociovanou s onemocněním. To je dáno velkou genetickou heterogenitou této klinické jednotky a možností výskytu změn v genech, které nejsou zavzaté do vyšetřovacího panelu. Příbuzní probandů, u kterých se nenašla genetická podstata jejich nemoci, by i při negativitě vstupního klinického vyšetření měli zůstat v pravidelném preventivním kardiologickém sledování. U dětí a mladých dospělých do 20 let s pozitivní rodinnou anamnézou doporučujeme klinické kontroly s nasnímáním EKG a echokardiografií jednou za 12–18 měsíců, u jedinců starších 20 let min. jednou za pět let. Doporučený vyšetřovací algoritmus je schematicky znázorněn na obr. 3.

Doručeno do redakce: 29. 9. 2016
Přijato po recenzi: 13. 10. 2016
MUDr. Linda Skutková
www.fnbrno.cz
Sources
1. Cahill TJ, Ashrafian H, Watkins H. Genetic cardiomyopathies causing heart failure. Circ Res 2013; 113(6): 660 – 675. doi: 10.1161/ CIRCRESAHA.113.300282.
2. Ashrafian H, Watkins H. Reviews of translational medicine and genomics in cardiovascular disease: new disease taxonomy and therapeutic implications cardiomyopathies: therapeutics based on molecular phenotype. J Am Coll Cardiol 2007; 49(12): 1251 – 1264.
3. Tfelt-Hansen J, Winkel BG, Grunnet M et al. Cardiac channelopathies and sudden infant death syndrome. Cardiology 2011; 119(1): 21 – 33. doi: 10.1159/ 000329047.
4. Ackerman MJ, Priori SG, Willems S et al. HRS/ EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Heart Rhythm 2011; 8(8): 1308 – 1339.
5. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17(5): 405 – 423. doi: 10.1038/ gim.2015.30.
6. Saj M, Bilinska ZT, Tarnowska A et al. LMNA mutations in Polish patients with dilated cardiomyopathy: prevalence, clinical characteristics, and in vitro studies. BMC Med Genet 2013; 14 : 55. doi: 10.1186/ 1471-2350-14-55.
7. Małek ŁA, Labib S, Mazurkiewicz Ł. A new c1621 C > G, p.R541G lamin A/ C mutation in a family with DCM and regional wall motion abnormalities (akinesis/ dyskinesis): genotype - phenotype correlation. J Hum Genet 2011; 56(1): 83 – 86. doi: 10.1038/ jhg.2010.137.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2016 Issue 4
Most read in this issue
- Dlouhodobé levokomorové srdeční podpory v léčbě srdečního selhání
-
Komentář k Doporučeným postupům ESC/ ČKS
Diagnostika srdečního selhání - Léčba hypertenze starších nemocných
- Lipertance® – dva problémy, jedno řešení