
Malobuněčná varianta světlobuněčného sarkomu měkkých tkání napodobující Ewingův sarkom - kazuistika
Authors:
Zdeněk Kinkor 1; Iveta Mečiarová 2; Petr Grossman 1; Tomáš Vaneček 1; Andrej Švec 3; Milan Kokavec 3
Authors‘ workplace:
Bioptická laboratoř s. r. o., Šiklův ústav patologie, LF UK, Plzeň
1; Alfa Medical Patológia, FN Ružinov, Bratislava
2; Ortopedická klinika, Univerzitná nemocnica Akademika Dérera, Bratislava
3
Published in:
Čes.-slov. Patol., 51, 2015, No. 1, p. 42-46
Category:
Original Article
Overview
Popisován je případ vzácné malobuněčné varianty světlobuněčného sarkomu měkkých tkání distálně na pravém boku u 42leté ženy. Nespecifická morfologie malobuněčného kulatobuněčného Ewing-like tumoru vedla původně k mylné interpretaci tumoru jako extraskeletální myxoidní chondrosarkom, Ewingův sarkom resp. maligní melanom. Typické rysy světlobuněčného sarkomu jako např. fascikulární vřetenobuněčné uspořádání, úprava buněk do hnízd či světlobuněčný resp. eozinofilní vzhled cytoplazmy nebyly zastiženy ani při extenzívním zpracování tkáně. Imunohistochemické vyšetření zjistilo expresi S100 proteinu a HMB45; markery CD99, FLI1, cytokeratiny a EMA byly negativní. Mikroskopický obraz primární léze, místní recidivy a následných mnohočetných kožních metastáz byl totožný. Molekulárně genetická analýza až ve vzdálené metastáze v kůži prokázala translokaci t(12;22)(EWSR1-ATF1) a nakonec vedla ke správné diagnóze neobvyklého světlobuněčného sarkomu měkkých tkání. Diskutován je význam využití genetických testů v diferenciální diagnostice s ohledem na existenci histogeneticky a biologicky rozdílných nádorů s analogickou molekulární alterací.
Klíčová slova:
světlobuněčný sarkom šlach a aponeuróz – Ewingův sarkom – Ewing-like – maligní melanom – malobuněčná varianta – t(12;22) (EWSR1-ATF1)
Světlobuněčný sarkom měkkých tkání/šlach a aponeuróz (SS) je vzácný, většinou několik let pomalu rostoucí, sarkom mladistvých a mladších dospělých (nejčastěji II. - IV. dekáda), vyskytující se v povrchových měkkých tkáních v intimní souvislosti šlach/fascií, zejména akrálních partií dolních (nárt, ploska, kotník) a horních (ruka, zápěstí) končetin (1-4). Přestože obvyklá morfologie signalizuje low-grade sarkom, pětileté přežití se uvádí jen kolem 50 % případů; nejčastější metastázování směřuje do regionálních lymfatických uzlin a plic (2-4). Histologická diagnostika je poměrně obtížná vzhledem k ojedinělosti výskytu, ale především proto, že „patognomonická“ světlobuněčnost tumoru je zde spíše výjimkou. SS je charakterizován blandně vyhlížejícími vřetenitými až oválnými buňkami s malým množstvím spíše eozinofilní cytoplazmy s výrazným jadérkem, uspořádanými do kompaktních snopců a hnízd oddělených různě silnými hyalinními vazivovými pruhy. Nezřídka jsou patrné rozptýlené obrovské vícejaderné elementy napodobující Toutonovy buňky resp. osteoklasty. Rozpoznání nádoru navíc komplikuje občasná přítomnost melaninového pigmentu a kompletně vyjádřený melanocytární fenotyp, včetně přesvědčivě dokumentované přítomnosti melanozomů na ultrastrukturální úrovni; to byl důvod pro dříve používané synonymické označení maligní melanom (MM) měkkých částí (1). Molekulární biologie posléze jednoznačně vyloučila histogenetickou příbuznost s maligním melanomem a prokázala translokace t(2;22) (EWSR1-CREB1) a t(12;22) (EWSR1-ATF1) jako zatím jediné známé klíčové patogenetické momenty (5,7).
Předkládáme neobvyklý případ malobuněčné varianty SS, který byl zpočátku mylně interpretován jako extraskeletální myxoidní chondrosarkom (EMCH), Ewingův sarkom (ES) resp. MM a teprve až při druhé recidivě/metastáze správně klasifikován molekulární analýzou.
MATERIÁL A METODIKA
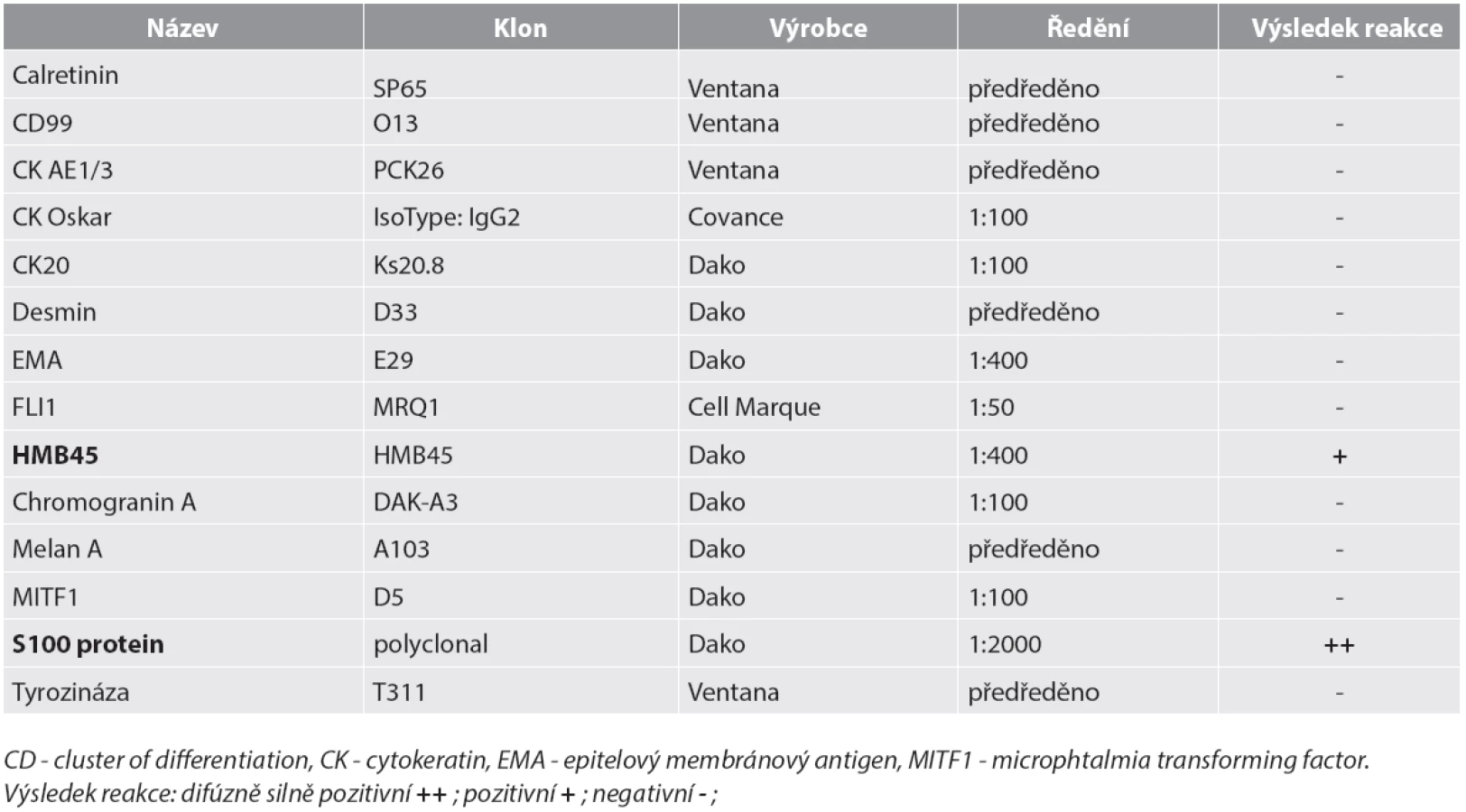
Materiál byl fixován v 10% formolu a zalit do parafínu (FFPE); pro barvení hematoxylinem eozinem a pro fluorescenční „in situ“ hybridizaci (FISH) byly krájeny řezy silné 2 mm, pro imunohistochemické vyšetření řezy silné 4 mm. Imunohistochemické vyšetření bylo prováděno elektronicky nastaveným protokolem v automatu BenchMark ULTRA, VENTANA/Roche. K vizualizaci reakce byl použit diaminobenzidin tetrahydrochlorid, k dobarvení jader metylénová modř. Seznam použitých protilátek, výrobce, klon a ředění jsou uvedeny v tabulce (tab. 1).
FISH analýza byla provedena prostřednictvím EWSR1 „break apart“ sondy (VYSIS/Abbott, Molecular, IL, USA) a následně též pomocí ATF1 a CREB1 „break apart“ sondy vyrobené z BAC (bakteriální arteficiální chromozom) klonů RP11-189H16 a RP11-407N8, resp. RP11-167C7 a RP11-381D21 (BlueGnome, Cambridge, UK). DNA byla izolována za použití QIAsymphony DNA Mini kitu (Qiagen, Hilden, Germany) na automatizovaném extrakčním systému QIAsymphony SP (Qiagen) dle protokolu výrobce pro FFPE vzorky (Purification of genomic DNA from FFPE tissue using the QIAamp DNA FFPE Tissue Kit and Deparaffinization Solution). RNA byla extrahována pomocí RecoverAll Total Nucleic Acid Isolation kitu (Ambion, Austin, TX, USA). Reverzní transkripce (RT) byla provedena kitem Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Mannheim, Germany). Mutace kodonu 600 (Val600) genu BRAF byla detekována pomocí PCR s FastStart PCR Master (Roche Diagnostic, Mannheim, Germany) a primery (BRAF-F: 5´ TCATGAAGACCTCACAGTAAAA 3´, BRAF-R: 5´ CAAAATGGATCCAGACAACT 3´) Výsledný PCR produkt byl analyzován přímým sekvenováním s Big Dye Terminator Sequencing kitem (Applied Biosystems, Foster City, CA, USA). Zároveň byla provedena reverzní hybridizace CE IVD certifikovaným kitem BRAF StripAssay (ViennaLab Diagnostics GmbH, Gaudenzdorfer Gürtel 43–45, A-1120 Vienna, Austria). K detekci fúzního transkriptu EWSR1-ATF1/CREB1 byla využita RT-PCR s HotStar Taq PCR Master Mixem (Qiagen) a s primery amplifikujícími oblast předpokládané fúze. Specifita fúzních transkriptů byla ověřena sekvenační analýzou (viz výše).
POPIS PŘÍPADU
U 42leté ženy byl zjištěn v měkkých tkáních distálně na pravém boku 50 mm veliký, ohraničený, neopouzdřený tumor, který byl patologem hodnocen jako EMCH. Po dvou letech došlo k místní recidivě a při shodné morfologii a expresi S100 proteinu uzavíráno týmž patologem opět jako EMCH. Za dalších 10 měsíců byla nalezena 20 mm veliká metastáza identického tumoru v měkkých tkáních třísla a dva jiní patologové dospěli k odlišným závěrům - jedná se buď o MM nebo ES/PNET resp. Askinův tumor s aberantním fenotypem. S odstupem několika následujících měsíců se objevily další (dle údajů klinika až kolem 20) kožní metastázy, především na trupu. Případ byl posléze konzultován na našem pracovišti a zpočátku byl rovněž jasně favorizován MM; dodatečně provedené molekulárně genetické vyšetření nakonec ukázalo, že se však jedná o zcela jiný nádor naprosto neodpovídající klasicky popisovanému histologickému obrazu.
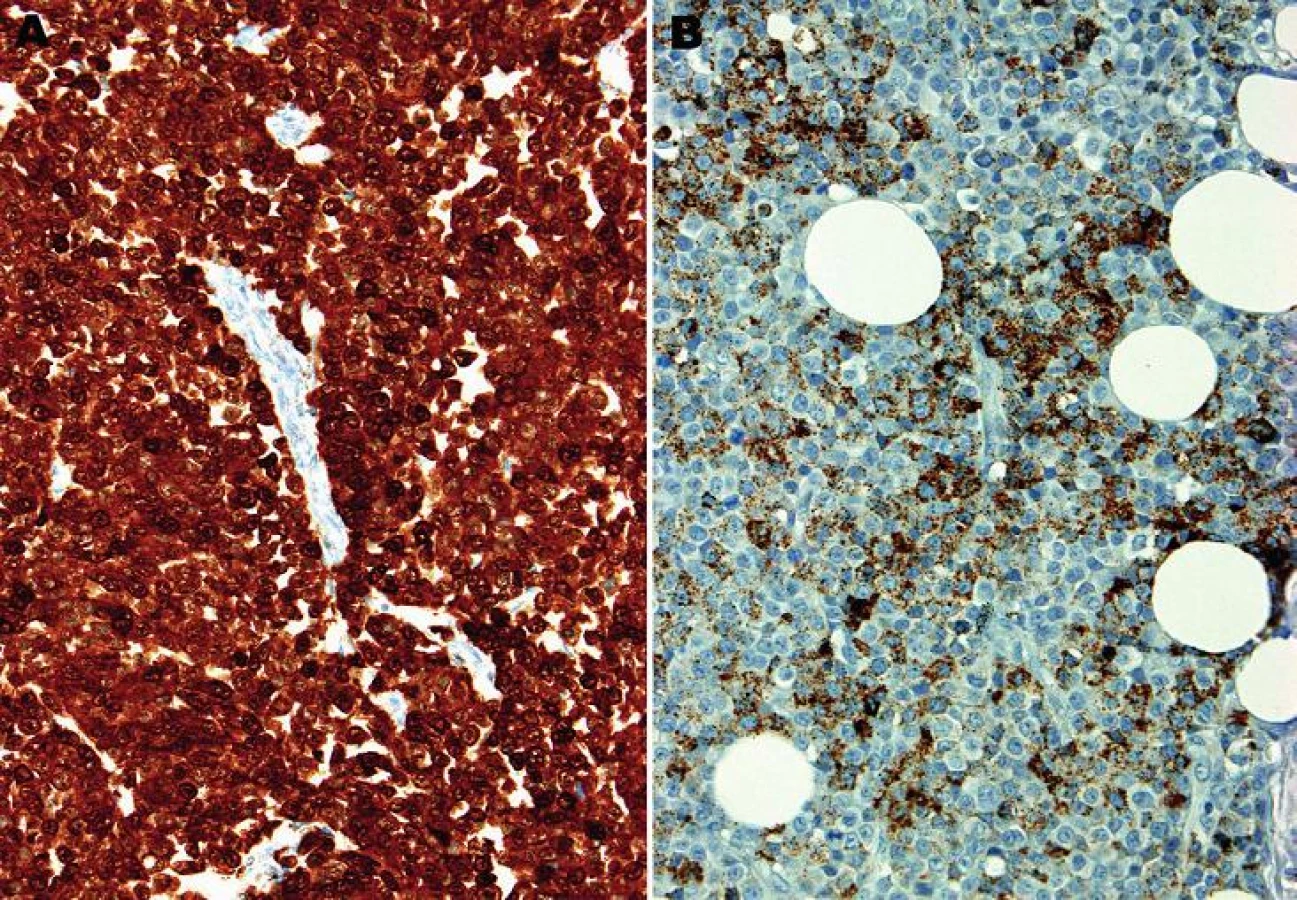
Mikroskopický vzhled spočíval v difúzní, monotónní mozaikovité, malobuněčné kulatobuněčné populaci odpovídající archetypu tzv. small blue round cell tumoru (obr. 1). Chybělo jakékoliv strukturální uspořádání (např. rozety) a částečně diskohezivní nádorové elementy s malým množstvím bazofilní cytoplazmy obsahovaly uniformní jádra s výraznými jadérky. Evidentní mitotická aktivita překračovala 20 mitóz na 10 velkých zorných polí (zvětšení 400x). Imunohistochemické vyšetření ukázalo difúzní silnou expresi S100 proteinu a ložiskově slaběji též HMB45 (obr. 2); další použité markery byly kompletně negativní (Melan A, tyrozináza, MITF, CD99, FLI1, cytokeratiny CK20, Oskar a AE1/3, EMA, chromogranin A, calretinin, desmin).

Molekulárně genetickým vyšetřením byl pomocí FISH metody prokázán zlom v oblasti genů EWSR1 a ATF1, nikoliv v oblasti genu CREB1 (obr. 3). Tyto nálezy byly konfirmovány detekcí fúzních transkriptů EWSR-ATF1 pomocí sekvenování (obr. 4). Současně s tím byla vyloučena také mutace V600E genu BRAF.


DISKUZE
Dlouhodobé systematické molekulárně biologické vyšetřování ukázalo, že na jedné straně mohou existovat histogeneticky, morfologicky i biologicky rozdílné nádory mající totožnou genetickou aberaci; na straně druhé tumory s jedinečným a naprosto odlišným molekulárním pozadím mohou být od sebe histologicky nerozeznatelné. Dokladem první z možností jsou např. translokace t(15;19) (ETV6-NTRK3) přítomná u kongenitálního fibrosarkomu, mezoblastického nefromu, mamárního resp. salivárního sekretorického karcinomu a akutní myeloidní leukémie nebo translokace t(2;22) (EWSR1-CREB1) a t(12;22) (EWSR1-ATF1) nacházející se u SS a jeho gastrointestinální analogie, angiomatoidního fibrózního histiocytomu (AFH), primárního plicního myxoidního sarkomu a světlobuněčného hyalinizujícího karcinomu slinných žláz (8-11). Vysvětlení pro takovou variabilitu výsledného projevu identické molekulární alterace zatím chybí, spekuluje se např. o významu výchozího tkáňového prostředí, ve kterém se genetická porucha odehraje (pojivová tkáň nebo žlázový epitel), nelze však vyloučit ani další, zatím neznámé genetické/epigenetické mechanizmy. Gen EWSR1 je ostatně velmi promiskuitní a translokačně značně potentní; dosud bylo nalezeno minimálně 15 rozdílných fúzních partnerství, která jsou ve svém důsledku generickým substrátem téměř stejného počtu rozličných neoplázií. V tomto ohledu skýtá obrovský potenciál nová technologie tzv. next generation sequencing, která umožní jak hlubší rozkrytí a pochopení složitého komplexu patogenetických dějů na úrovni DNA i RNA (transkriptomu), tak hledání optimálních míst pro cílenou (targeted) léčbu.
Ukázkou druhé eventuality je škála tzv. small blue round cell tumorů, kde neodlišitelný histologický obraz a částečně překrývající se fenotyp již bez cíleného molekulárního testování přesnou diagnózu neumožňují. Omezíme-li se pouze na zástupce měkkotkáňových lézí, lze vedle ES/PNET (včetně heterogenní skupiny tzv. Ewing family tumorů) a desmoplastického malobuněčného kulatobuněčného tumoru zmínit např. i nediferencovaný alveolární rhabdomyosarkom, nediferencovaný synoviální sarkom, nediferencovaný EMC, primární myoepiteliom/myoepiteliální karcinom atd. Nově sem lze zcela jistě přiřadit nedávno popsané, čistě molekulárně definované, sarkomy s translokací t(4;19) (CIC-DUX4) a inv(X)(p11.4p11.22) (BCOR-CCNB3) (12,13).
Naše raritní pozorování malobuněčné Ewing-like varianty SS je jen dalším důkazem možné proměnlivosti mikroskopického obrazu u léze s jasně vymezeným genetickým profilem. Konzistentní „melanomový“ fenotyp SS je v tomto kontextu značně zavádějící a znesnadňuje správné rozpoznání tumoru, neboť právě MM dokáže dokonale napodobit prakticky jakýkoliv nádor. V literatuře jsme zaznamenali pouhé dvě práce dokumentující malobuněčnou verzi SS napodobující ES - a to v měkkých tkáních (lýtko) a primárně ve skeletu (kost stydká) (14,15). Klasifikace prvního ze jmenovaných případů je však dosti problematická; tumor totiž difúzně exprimoval pouze desmin; markery S100 protein a HMB45 byly kompletně negativní. Je otázkou, zda se ve skutečnosti nejednalo o malobuněčnou variantu AFH, kde právě častá exprese desminu může být při odpovídající morfologii důležitým vodítkem ke správné diagnóze; pochopitelně následně podpořené molekulární analýzou. Obě publikace se shodují v názoru, že bez extenzívních molekulárních testů by léze zůstaly nerozpoznány a byly by reportovány jako ES/PNET.
Náš případ mělo možnost hodnotit několik zkušených patologů a spektrum diagnóz zahrnovalo EMC, ES/PNET a MM; pro nás byla při druhém čtení diagnóza melanomu zpočátku též nejpřijatelnější. Prakticky jen z určité zvědavosti jsme provedli dvě orientační genetická vyšetření s překvapivým výsledkem - mutace V600E genu BRAF negativní; metodou FISH zjištěn zlom genu EWSR1. Posléze již jediné cílené vyšetření prokázalo translokaci t(12;22) (EWSR1-ATF1). Konečná diagnóza histologicky mimořádně vzácného SS pak byla nepochybná.
V této souvislostí nelze nepřipomenout ještě vzácnější viscerální analogii SS uváděnou v písemnictví jako clear cell sarcoma-like tumor with osteoclast-like giant cells či nověji jako gastrointestinal neuroectodermal tumor (GNET). Dosud není shoda na tom, zda se jedná o totožný/příbuzný tumor či úplně jinou lézi (16,18). Každopádně GNET (většinou lokalizovaný v ileu a mikroskopicky mimo jiné nepříjemně kopírující např. GIST či karcinoid/epitelový NET) při expresi S100 proteinu reaguje konzistentně negativně s dalšími melanocytárními markery (melan A, HMB45, tyrozináza, MITF1), postrádá melaninový pigment a známky melanogeneze na ultrastrukturální úrovni. Obráceně, GNET často pozitivně reaguje s antigeny SOX10, NSE, synaptofyzin, neurofilamenta) a v ultrastruktuře vykazuje sekreční vezikuly a denzní granula. Genotypově GNET charakterizuje účast CREB1 jako preferenčního translokačního fúzního partnera genu EWSR1 (19,20). Jakkoliv není zatím souvislost mezi SS a GNET objasněna, je nejdůležitější nezaměnit oba dva za MM. Při nálezu domnělých metastáz MM do zažívacího traktu, zejména při absenci primární kožní léze, je nutné tuto vzácnou diagnostickou eventualitu bezpečně vyloučit.
ZÁVĚR
Popisovaný případ morfologicky mimořádně vzácné, malobuněčné varianty SS dokládá nutnost využití molekulární biologie v rutinní diagnostické praxi. Variabilita histologického obrazu může vést k tomu, že i léze se zdánlivě přesvědčivě doloženým fenotypem, odpovídajícím kontextu „hematoxylin eozinové“ morfologie, může uniknout správnému rozpoznání. Dnes již běžně dostupná, byť cíleně zaměřená FISH, nemusí dát ale vždy definitivní odpověď (např. při zlomu genu EWSR1) a někdy je potřeba přesně určit i konkrétního partnera výsledného fúzního genu. Patolog pak musí alespoň teoreticky tyto souvislosti vnímat v diferenciální diagnostice.
DODATEK
Dodatečně bylo zjištěno, že pacientka zemřela na systémovou diseminaci sarkomu za 39 měsíců od primární manifestaci nádoru.
PODĚKOVÁNÍ
Autoři děkují prim. MUDr. Petrovi Mukenšnáblovi, Ph.D. za zhotovení fotodokumentace a Mgr. Vratislavu Šedivcovi za laboratorně technologickou a grafickou asistenci.
Adresa pro korespondenci:
Doc. MUDr. Zdeněk Kinkor, Ph.D.
Bioptická laboratoř s.r.o., Mikulášské nám. 4
326 00 Plzeň
tel.: 737 220 449
e-mail: kinkor@medima.cz
Sources
1. Hucks MJ, Saldivar VA, Chintagumpala MM et al. Malignant melanoma of soft parts involving the head and neck region: review of the literature and case report. Ultrastruct Pathol 1995; 19 : 395-400.
2. Deenik W. Mooi WJ, Rutgers EJ, Peterse JL et al. Clear cell sarcoma (malignant melanoma) of soft parts: a clinicopathologic study of 30 cases. Cancer 1999; 86 : 969-975.
3. Finley JW, Hanypsiak B, McGrath B, Kraybill W, Gibbs J. Clear cell sarcoma: the Roswell Park experience. J Surg Oncol 2001; 77 : 16-20.
4. Ferrari A, Casanova M, Bisogno G, Mattke A. Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German Soft tissue sarcoma cooperative group. Cancer 2002; 94 : 3269-76.
5. Panagopoulos I, Mertens F, Debiec-Rychter M, Isaksson M, et al. Molecular genetics characterization of the EWS/ATF1 fusion gene in clear cell sarcoma of tendons and aponeuroses. Int J Canc 2002; 99 : 560-567.
6. Hisaoka M, Ishida T, Kuo TT, Matsuyama A et al. Clear cell sarcoma of soft tissue: a clinicopathologic, immunohistochemical and molecular analysis of 33 cases. Am J Surg Pathol 2008; 32 : 452-460.
7. Wang WL, Mayordomo E, Zhang W, Hernandez VS et al. Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcript in clear cell sarcoma (melanoma of soft parts). Mod Pathol 2009; 22 : 1201-1209.
8. Sheng WQ, Hisaoka M, Okamoto S, Tanaka A et al. A clinicopathologic study of 10 cases and molecular detection of the ETV6-NTRK3 fusion transcripts using paraffin-embedded tissues. Am J Clin Pathol 2001; 225 : 348-355.
9. Skálová A, Vaněček T, Šíma R, Laco J et al. Mammary analogue secretory carcinoma of salivary gland containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland entity. Am J Surg Pathol 2010; 34 : 599-608.
10. Kralik JM, Kranewitter W, Boesmueller H, Marschon R et al. Characterization of a newly indentified ETV6-NTRK3 fusion transcript in acute myeloid leukemia. Diagn Pathol 2011; 6 : 19-23.
11. Thway K, Fisher C. Tumors with EWSR1-CREB1 and EWSR1-ATF1 fusions: the current status. Am J Surg Pathol 2012; 36: e1-11.
12. Graham C, Chilton-Macneill S, Zielenska M, Somers GR. The CIC-DUX4 fusion transcript is present in a subgroup of primitive pediatric round cell sarcomas. Hum Pathol 2012; 43 : 180-189.
13. Pierron G, Tirode F, Lucchesi C, et al. A new subtype of bone sarcoma defined by BCOR-CCNB3 gene fusion. Nat Genet 2012; 44 : 461-466.
14. Pacheco M, Horsman DE, Haves MM, Clarkson PW et al. Small blue round cell tumor of the interosseous membrane bearing a t(2;22)(EWSR1-CREB1) translocation: a case report. Mol Cytogenet 2010; 3 : 12.
15. Nakayama S, Yokote T, Iwaki K, Akioka T et al. A rare case of primary clear cell sarcoma of the pubic bone resembling a small round cell tumor: an unusual morphologic variant. BMC Cancer 2012; 12 : 538.
16. Friedrichs N, Testi MA, Moiraghi L, Modena P et al. Clear cell sarcoma-like tumor with osteoclast-like giant cells in the small bowel: further evidence for a new tumor entity. Int J Surg Pathol 2005; 13 : 313-318.
17. Antonescu CR, Nafa K, Segal NH, Dal Cin P, Landanyi M. EWS-CREB1: a recurrent fusion in the clear cell sarcoma-association with gastrointestinal location and absence of melanocytic differentiation. Clin Canc Res 2006; 12 : 5356-5362.
18. Lyle PL, Amato CM, Fitzpatrick JE, Robinson WA. Gastrointestinal melanoma or clear cell sarcoma? Molecular evaluation of 7 cases previously diagnosed as malignant melanoma. Am J Surg Pathol 2008; 32 : 858-866.
19. Kosemehmetoglu K, Folpe AL. Clear cell sarcoma of tendons and aponeuroses and osteoclast-rich tumor of the gastrointestinal tract with features resembling clear cell sarcoma of soft parts: a review and update. J Clin Pathol 2010; 63 : 416-423.
20. Stockman DL, Miettinen M, Suster S, Spagnolo D et al. Malignant gastrointestinal neuroectodermal tumor: clinicopathologic, immunohistochemical, ultrastructural and molecular analysis of 16 cases with a reappraisal of clear cell sarcoma-like tumor of the gastrointestinal tract. Am J Surg Pathol 2012; 36 : 857-868.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2015 Issue 1
Most read in this issue
- Neurofibromatosis von Recklinghausen typ 1 (NF1) – klinický obraz a molekulárně-genetická diagnostika
- Patologické hodnocení vzorků kolorektálního karcinomu: pokročilé a časné léze
- Fibroadenom prsu s pleomorfními stromálními buňkami
- Malobuněčná varianta světlobuněčného sarkomu měkkých tkání napodobující Ewingův sarkom - kazuistika