
Marshall and stickler syndrome in one family
Authors:
D. Tomčíková 1; B. Bušányová 1; V. Krásnik 2; A. Gerinec 1
Authors‘ workplace:
Klinika detskej oftalmológie NÚDCH - LFUK Bratislava, Limbová 1, 83340 Bratislava., Prednosta: MUDr. Dana Tomčíková, PhD, MHA.
1; Klinika oftalmológie UNB a LFUK Bratislava, Ružinovská 6, 82606 Bratislava., Prednosta: doc. MUDr. Vladimír Krásnik, PhD.
2
Published in:
Čes. a slov. Oftal., 74, 2018, No. 3, p. 108-111
Category:
Case Report
doi:
https://doi.org/10.31348/2018/1/5-3-2018
Overview
The authors present the ocular finding in a patient sent to the Department of Paediatric Ophthalmology at the Children's University Hospital – Faculty of Medicine, Comenius University in Bratislava at the age of 3 months, with congenital glaucoma in her right eye and bilateral high myopia. The family anamnesis of the patient shows repeated occurrence of stunted growth, myopia, facial dysmorphia and cataract. The child's mother had high myopia, the mother's brother underwent cataract surgery, the child's grandmother and her sisters and the child's great grandmother had high myopia and glaucoma, and probably underwent cataract surgery at a young age. The child's mother and grandmother underwent a genetic examination, with a conclusion of Marshall syndrome. Within the framework of neonatal screening, poor cortical auditory evoked potential, a defect of the interventricular septum and bifid uvula were diagnosed in the child. With regard to the overall finding in the patient and the genetic family history, we suspected Marshall syndrome. A genetic examination determined Stickler syndrome type 1 with the presence of mutation in the COL2A gene (variant c.2710C >T (p.Arg904Cys,rs121912882)). Due to high intraocular pressure with the impossibility of compensation by medication, bilateral trabuculectomy was performed on the patient. At present the patient has intraocular pressure compensated with adjuvant medicamentous therapy. With regard to high myopia and pronounced degenerative changes on the periphery of the retina in the sense of lattice degeneration, preventive cryopexy of the retinal periphery is planned. A molecular genetic analysis helped diagnose the pathology as Stickler syndrome type 1, which manifested phenotype symptoms of Marshall syndrome or Stickler syndrome type 2.
Key words:
Marshall syndrome, Stickler syndrome, mid-facial dysmorfism, myopia, glaucoma, cataract.
Introduction
Marshall syndrome is a genetically conditioned heterogeneous pathology characterised by high myopia, cataract, glaucoma, retinal detachment (amotio retinae), progressive hearing defect, mild spondyloepiphyseal dysplasia, stunted growth, osteoarthritis, central facial dysmorphia and cleft palate. The intensity of the individual symptoms varies between individuals, phenotype manifestations characteristic of Marshall syndrome overlap with Stickler syndrome. Originally 3 types of Stickler syndrome were differentiated, nosologically separated from Marshall syndrome. In Stickler syndrome there is more frequent incidence of hyperflexibility of joints, while growth disorders are not pronounced. Hearing impairments frequently occur in Stickler syndrome types 2 and 3, in type 1 they are not present. Ocular symptoms do not occur in Stickler syndrome type 3. The incidence of the pathology is 1 : 7500-9000 newborn infants. The syndrome may be hereditary, autosomal dominant or recessive (1, 6, 7, 8). Abnormal synthesis of collagen type II, XI or IX, located in cartilage, vitreous body, intervertebral discs and inner ear is present in patients. Marshall syndrome is conditioned by the mutation of gene COL11A1. In Stickler syndrome mutations of COL2A1, COL11A2, COL9A1, COL9A2, COL9A3 and COL11A1 occur. Phenotype differences between Marshall syndrome and Stickler syndrome are stated in the case of Marshall syndrome as stunted growth, low root of nose, early onset of hearing disorder and more pronounced maxillary hypoplasia. In Stickler syndrome the growth defects become less pronounced with age (3). Heredity is autosomal dominant (COL2A1, COL11A2, COL11A1) and recessive (COL9A1, COL9A2, COL0A3). In the past Marshall syndrome and Stickler syndrome were considered different pathological units with phenotypically similar manifestations. With regard to the discovery of the mutation on the same locus in both syndromes (Marshall syndrome and Stickler syndrome type II – COL11A1), it is necessary to cease to separate these 2 syndromes (6, 7, 4).
Method
The patient was sent to the Department of Paediatric Ophthalmology of the Children's University Hospital – Faculty of Medicine, Comenius University in Bratislava in 2017 at the age of 3 months due to pain and lachrymation in the right eye. Within the framework of an ophthalmological examination, the following were tested: central visual acuity (CVA), echobiometry. Due to the patient's age, we examined intraocular pressure (IOP), gonioscopy, local finding on anterior segment, optical media, ocular fundus and refraction under general anaesthesia (GA). The patient's CVA was examined with the aid of the method of preferential looking. For the examination of the anterior segment we used direct examination by surgical microscope and manual slit lamp. We measured IOP on an I care instrument. We examined the gonioscopic finding with the aid of a three-sided Goldmann lens. The finding on the ocular fundus of the patient was examined by indirect ophthalmoscope in artificial mydriasis. We also measured refraction in artificial mydriasis, using a Retinomax manual keratorefractometer. For photo-documentation we used a RetCam II. In addition to the ophthalmological examination, biochemical and genetic tests were also conducted.
Results
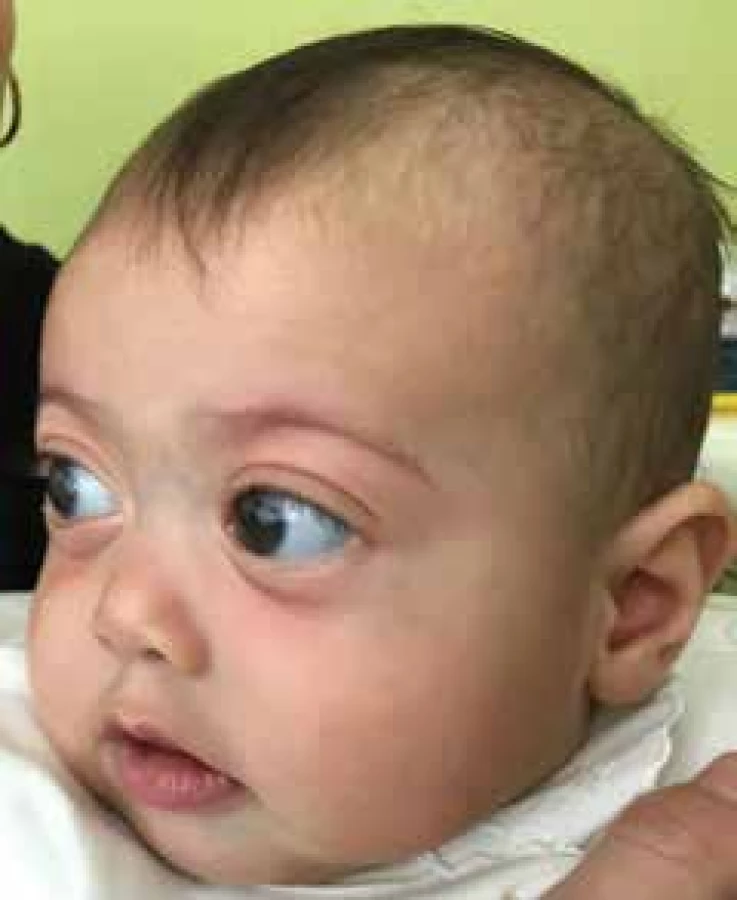
At the examination congenital glaucoma was diagnosed in the right eye. Due to concurrent infection of the upper respiratory tracts, the examination under general anaesthesia was deferred for 2 weeks, local antiglaucomatous therapy was commenced in a triple combination of antiglaucomatous agents Cosopt and Brimonal 0.2%. At the examination under GA we determined pseudoexophthalmos in the right eye, with slightly increased diameter of cornea. The cornea was edematous, anterior chamber deeper, pupil 4 mm wide, with delayed reaction to illumination. The optical media were clear. The finding on the ocular fundus was more difficult to evaluate due to corneal edema. In the left eye the finding was physiological. Other numerical values are stated in table no. 1. A positive family anamnesis was present (fig. 1). High myopia and hearing disorder were diagnosed in the mother, and later Marshall syndrome was genetically confirmed. The mother's brother had undergone cataract surgery, the child's grandmother, her sisters and the child's great-grandmother had high myopia, cataract and hearing impairment. All the relatives had stunted growth. Within the framework of neonatal screening, poor cortical auditory evoked potential, bifid uvula and a defect of the interventricular septum were diagnosed in the child. With regard to the positive family anamnesis, the presence of glaucoma, high angle myopia and characteristic facial dysmorphia, we indicated a genetic examination for suspected Marshall syndrome, as well as genetic examination for the presence of the CYP1B1 gene, which is characteristic of primary congenital glaucoma. Due to insufficient medicamentous compensation of IOP, trabulectomy was performed on the right eye in February 2017. At a postoperative follow-up examination in March 2017, increased intraocular pressure with slight corneal edema was determined also in the left eye. In the right eye, following regression of corneal edema, Haab's striae became visible, and later appeared also in the left eye. Excavation on the disc of the optic nerve (DON) remained 0.2 in the right eye, in the left eye DON without excavation (fig. 4, 5). A genetic examination for the presence of the CYP1B1 gene was negative. An audiometric examination confirmed a perceptual hearing disorder in the child on the level of 31% reduction bilaterally. A defect of the interventricular septum spontaneously healed. An orthopaedic examination did not detect any pathology. Central facial dysplasia and cleft palate were present in the child (fig. 2, 3). A genetic examination detected mutation of COL2A1 (variant c.2710C >T (p.Arg904Cys,rs121912882)), which is typical of Stickler syndrome type 1. Upon an examination at the age of 1.5 years the patient's condition was stabilised upon local anti-glaucomatous therapy with Cosopt gtts bilaterally and Xalatan gtts in the right eye (fig. 6). With regard to the risk of retinal detachment with the existing degenerative changes of the retinal periphery upon high myopia, we are planning preventive cryoretinopexy (5). The numerical values at the age of 15 months are presented in table no. 2.







Discussion
Marshall/Stickler syndrome is characterised by a disorder of the conjunctival tissue. The presence of abnormal synthesis of collagen type II, XI or IX, which are located in the hyaline cartilage, vitreous body, intervertebral discs and inner ear explains the multisystemic affliction of the patients. The clinical picture often varies, the individual symptoms of the syndrome are not equally developed in all patients. In patients with the phenotype of Marshall/Stickler syndrome but without demonstrated mutation in the region of the COLA11A1 gene, blue sclera and keratoglobus have also been described (4). Mutation in the COLA11A1 gene causes a disorder in the synthesis of collagen IX, mutation in the gene COL2A1 is more typical of the phenotype of Stickler syndrome and codes collagen II. According to the intensity of the disorder, varying degrees of defect of the conjunctival tissue are present. Spontaneous rupture of the lens capsule has been described in a child during a glaucomatous seizure (2). With regard to these serious, sometimes crippling consequences of this syndrome, active monitoring is essential due to the most frequent known complications.
Conclusion
Marshall/Stickler syndrome involves severe, sometimes crippling consequences. Due to the known risks and complications, it is essential to monitor the patient actively, especially from an ophthalmological perspective – compensation of IOP, monitoring and accurate correction of myopia, active monitoring of peripheral retinal degeneration. In the case of pronounced changes on the retinal periphery it is necessary to indicate preventive retinopexy. The patient and parents must be instructed with regard to self-monitoring for the purpose of timely identification of potential retinal detachment. It is also necessary to monitor the patient with regard to the potential development of cataract. Monitoring by an ophthalmologist is essential also from the perspective of preserving the most optimal visual functions, since the patient is also affected by hearing impairment. The patient therefore requires active monitoring also with an ENT specialist and orthopaedic surgeon. No less important is genetic consultation in the affected family with regard to further children. Our patient is being regularly monitored for all of the above-stated complications. At present her condition is stabilised.
The authors of the study declare that no conflict of interest exists in the compilation, theme and subsequent publication of this professional communication, and that it is not supported by any pharmaceuticals company.
MUDr. Dana Tomčíková, PhD, MHA

Klinika detskej oftalmológie NÚDCH - LFUK Bratislava
Limbová 1
83340 Bratislava
Sources
1. Annunen, S., Körkkö, J., Czarny, M.: Splicing Mutations of 54-bp Exons in the COL11A1 Gene Cause Marshall Syndrome, but Other Mutations Cause Overlapping Marshall/Stickler Phenotypes. Am J Hum Genet., 65(4); 1999 : 974–983.
2. Endo, S., Hashimoto, Y., Ishida, N.: A case of Marshall syndrome with secondary glaucoma due to spontaneous rupture of the lens capsule. Nippon Ganka Gakkai Zasshi., 102(1); 1998 : 75-9.
3. Imamoglu, S., Kaya, V., Imamoglu, E., Y.: Congenital keratoglobus with blue sclera in two siblings with overlapping Marshall/Stickler phenotype. Indian J Ophthalmol. 64(11); 2016 : 856–859.
4. Higuchi, Y., Hasegawa, K., Yamashita, M.: A novel mutation in the COL2A1 gene in a patient with Stickler syndrome type 1: a case report and review of the literature.: J Med Case Rep. 2017; 11 : 237.
5. Karel I., Doležalová J., Oudová P.: Stickleruv syndrom (Dystrophia vitreoretinalis hereditaria). Výsledky operace odchlípení sítnice. Čes a slov. Oftal., 2001, (3),p. 147-154
6. Khalifa, O., Imtiaz, F., Ramzan, K.: Marshall syndrome: further evidence of a distinct phenotypic entity and report of new findings. Am J Med Genet A., 164A(10); 2014 : 2601-6.
7. Sakka, R., Kerkeni, E., Chaabouni, M.: Marshall syndrome: Clinical, radiological and genetical features of a Tunisian family. Tunis Med., 93(3); 2015 : 170-4.
8. Wright, K.W., Spiengel, P. H.: Pediatric Ophthalmology and Strabismus, Springer, New York 2003, p.1021-1058.
Labels
OphthalmologyArticle was published in
Czech and Slovak Ophthalmology

2018 Issue 3
Most read in this issue
- Ophthalmic manifestations of acute leukaemias
- Asphercial iols and their effect on visual, depth of field, spherical aberration and contrast sensitivity
- Congenital central toxoplasmic chorioretinitis - case study
- Marshall and stickler syndrome in one family