
Schnitzlerovej syndróm – kazuistická séria
Authors:
M. Bajer 1,2; L. Kapustová 3,4; S. Puteková 1,2; S. Blažíčková 2; M. Jeseňák 3,4
Authors‘ workplace:
Klinika vnútorného lekárstva FN Trnava a SZU a TU, Slovensko
1; FZaSP, TU v Trnave, Slovensko
2; Klinika detí a dorastu, Jesseniova lekárska fakulta v Martine, Univerzita Komenského v Bratislave, Univerzitná nemocnica Martin, Slovensko
3; Ústav klinickej imunológie a lekárskej genetiky, Jesseniova lekárska fakulta v Martine, Univerzita Komenského v Bratislave
4
Published in:
Klin Onkol 2025; 38(2): 119-127
Category:
Case Reports
doi:
https://doi.org/10.48095/ccko2025119
Overview
Východiská: Schnitzlerovej syndróm je získané autoinflamačné ochorenie so základom v poruche prirodzenej imunity. Podozrivým je proteín MyD88 – toll-like receptor zapojený do zápalovej kaskády vyúsťujúcej aj do zvýšenej sekrécie interleukínu-1 (IL-1), kľúčového cytokínu v patogenéze a klinickej manifestácii viacerých autoinflamačných stavov. Syndróm je pomenovaný po francúzskej kožnej lekárke Liliane Schnitzler, ktorá v roku 1972 opísala kazuistickú sériu pacientov s prejavmi urtikárie v spojení s monoklonálnou gamapatiou. V klinickom obraze dominujú prejavy chronickej urtikárie, pričom histologicky ide najčastejšie o neutrofilovú dermatózu. Nevyhnutným kritériom na zadefinovanie Schnitzlerovej syndrómu je monoklonálna gamapatia, pričom až v 90 % prípadov ide o monoklonálnu IgM gamapatiu. Zvyšná skupina pacientov má prítomnú gamapatiu v triede IgG. Z vedľajších kritérií potrebných na potvrdenie syndrómu podľa Strassburgskej klasifikácie je to zvýšená nešpecifická zápalová aktivita (elevácia sérového CRP, leukocytóza), morfologické zmeny na kostiach (verifikované hyperostotické či osteosklerotické zmeny skeletu na CT, scintigraficky, alebo PET/CT s použitím rádioaktívneho natrium fluoridu NaF18), bolesti kostí či artralgia. Pacienti sú vo zvýšenom riziku rozvoja lymfoproliferačného ochorenia ako Waldenströmovej makroglobulinémie alebo low-grade lymfómu (15–20 %). Rizikom je tiež rozvoj AA amyloidózy pri dlhodobej nekontrolovanej hyperinflamácii. Akútnou, život ohrozujúcou komplikáciou môže byť syndróm aktivovaných makrofágov. Vzhľadom na imunologickú podstatu ochorenia a eleváciu zápalových cytokínov je základom anticytokínová liečba. Najlepšie výsledky sú pozorované pri anakinre (antagonista receptora pre IL-1), inou možnosťou je eventuálne kanakinumab (monoklonálna protilátka proti IL-1b). Pozitívny efekt bol opisovaný aj pri inhibítoroch Brutonovej tyrozínkinázy. Kortikoidy a konvenčné imunosupresíva nie sú dostatočne efektívne. Prípad: V predkladanom texte prezentujeme formou kazuistickej série dve pacientky so Schnitzlerovej syndrómom s raritnejšími klinickými príznakmi a s diferencovanou odpoveďou na anticytokínovú liečbu. Cieľom autorov bolo informovať a zlepšiť povedomie o tomto vzácnom ochorení a jeho možnostiach liečby. Záver: Ochorenie je chronické, liečba je len symptomatická, ale vedie k ústupu klinických príznakov a dosiahnutiu kontroly nad zápalom. Riziko vzniku hematologickej malignity anticytokínová liečba pravdepodobne neovplyvňuje.
Klíčová slova:
autoimunita – autoinflamácia – monoklonálna gamapatia – Schnitzlerovej syndróm – urtikária
Úvod
Schnitzlerovej syndróm je chronické ochorenie spojené s nadmerným zápalovým procesom. Radí sa medzi získané autoinflamačné ochorenia s pravdepodobným základom v poruche regulácie prirodzenej imunity. Podozrivým je proteín MyD88 – toll-like receptor (TLR) zapojený do zápalovej kaskády vyúsťujúcej aj do zvýšenej sekrécie interleukínu-1 (IL-1), kľúčového cytokínu v patogenéze viacerých autoinflamačných ochorení. Autoinflamácie svojim klinickým obrazom často pripomínajú klasické autoimunitné ochorenia, avšak bez detekcie špecifických či nešpecifických autoprotilátok. Podstatou autoinflamačných ochorení je dysregulovaná zápalová kaskáda a mechanizmy vrodenej imunity. Samotné autoinflamačné ochorenia boli opísané prvý krát v roku 1999, kedy pracovná skupina okolo McDermotta diagnostikovala tumor necrosis factor receptor-associated periodic syndrome (TRAPS). Popísali tak patofyziologický mechanizmus systémového zápalového ochorenia pri absencii autoimunitných či iných známych mechanizmov, ktoré by ochorenie vyvolali a uviedol sa pojem autoinflamácia [1]. Úplne prvým ochorením z tohto spektra bola spomenutá v literatúre familiárna stredomorská horúčka (FMF), zmienená už v roku 1945 [2], ktorá je zároveň aj najčastejšou autoinflamáciou. V českej literatúre prvú publikáciu o Schnitzlerovej syndróme evidujeme z roku 2008, kde autori opisujú prípad pacienta po 14 rokoch trvajúcej diagnostike. Ako prvý v Českej republike bol tento pacient od roku 2007 liečený anakinrou [3]. Publikáciu od slovenských autorov zatiaľ neevidujeme. Schnitzlerovej syndróm je pomenovaný po francúzskej kožnej lekárke Liliane Schnitzler, ktorá v roku 1972 opísala kazuistickú sériu pacientov s prejavmi urtikárie v spojení s detekciou monoklonálnej gamapatie. V klinickom obraze dominujú prejavy chronickej urtikárie (eventuálne skôr urticaria-like prejavy, vzhľadom na abscenciu pruritu sprevádzajúceho klasické formy urtikárie), pričom histologicky ide najčastejšie o neutrofilovú dermatózu. Nevyhnutným kritériom na potvrdenie Schnitzlerovej syndrómu je monoklonálna gamapatia, pričom až v 90 % prípadov ide o monoklonálnu IgM gamapatiu. Zvyšná skupina pacientov má prítomnú gamapatiu v triede IgG. Z vedľajších kritérií potrebných na potvrdenie diagnózy Schnitzlerovej syndrómu podľa Strassburgskej klasifikácie (tab. 1) je prítomná u pacientov ešte zvýšená nešpecifická zápalová aktivita (elevácia sérového CRP, leukocytóza), bolesti kostí či artralgie a hyperostotické či osteosklerotické zmeny na kostiach, prípadne v kombinácii s osteolytickými zmenami. Kostné zmeny je nutné verifikovať pomocou CT vyšetrenia, ktoré je senzitívnejšou metódou ako klasické RTG. Možnosťou je scintigrafia skeletu s použitím technecium pyrofosfátu, najsenzitívnejšou metódou sa javí detekcia novotvorby osteoidu a hydroxyapatitu pomocou PET/CT zobrazenia s použitím rádioaktívneho natrium fluoridu NaF18 ako rádiofarmaka, nie však klasické FDG-PET/CT. Pacienti sú vo zvýšenom riziku rozvoja lymfoproliferačného ochorenia, najmä Waldenströmovej makroglobulinémie alebo low-grade lymfómu (15–20 %). Spojenie medzi Schnitzlerovej syndrómom a Waldenströmovou makroglobulinémiou je zaujímavé. Až 90 % pacientov s Waldenströmovou makroglobulinémiou vykazuje somatickú mutáciu L265P v géne MYD88. Ak je táto mutácia prítomná v B lymfocytoch, riadi rozvoj lymfoidnej malignity cez toll-like receptorovú signalizačnú cestu. MYD88 je tiež riadiacim proteínom pri aktivácii receptora pre IL-1 [4]. Uvedená mutácia bola opísaná u 9 z 30 pacientov so Schnitzlerovej syndrómom, takže sa nezdá byť jediným mechanizmom vzniku ochorenia [5]. Iným rizikom pri dlhodobej nekontrolovanej hyperinflamácii je tiež rozvoj AA-amyloidózy, pričom sa ukladá amylopeptid A, čo môže súvisieť aj s odďaľovaním nasadenia správnej liečby pri nesprávnej diagnóze. Zaujímavé je, že amyloidóza ľahkých reťazcov – primárna amyloidóza, ktorá je spojená s mnohopočetným myelómom, lymfómom alebo Waldenströmovou makroglobulinémiou, nebola zaznamenaná u pacientov so Schnitzlerovej syndrómom, napriek pretrvávaniu monoklonálnej gamapatie v priebehu ochorenia počas liečby [6]. Akútnou, život ohrozujúcou komplikáciou, ako uvádzame aj u našej pacientky, môže byť syndróm aktivovaných makrofágov (macrophage activation syndrome – MAS). Ide o závažný hyperinflamačný stav, ktorý môže komplikovať viacero stavov, vrátane autoinflamačných ochorení. Vzhľadom na imunologickú podstatu ochorenia a eleváciu zápalových cytokínov – najmä IL-1 či IL-6, je základom liečby u pacientov anticytokínová liečba. Najlepšie výsledky sú pozorované pri anakinre (antagonista receptora pre IL-1), inou možnosťou je eventuálne kanakinumab (monoklonálna protilátka proti IL-1b). Pozitívny efekt bol opisovaný aj pri ibrutinibe (inhibítor Brutonovej tyrozínkinázy). Kortikoidy a konvenčné imunosupresíva ako cyklofosfamid či cyklosporín nie sú dostatočne efektívne, pri miernom priebehu ochorenia môže mať efekt kolchicín [7]. Ochorenie je chronické, liečba je len symptomatická, ale vedie k ústupu klinických príznakov. Riziko vzniku hematologickej malignity anticytokínová liečba pravdepodobne neovplyvňuje.
![Strassburgské kritériá pre Schnitzlerovej syndróm [19].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/7c87b978bb48c291803f83167828fc08.png)
Cieľ a metóda
Formou prvej kazuistiky prezentujeme ojedinelý klinický prípad pacientky, u ktorej sa Schnitzlerovej syndróm manifestuje závažným hyperinflamačným stavom a časový odstup od prvých príznakov po správne stanovenie diagnózy a efektívnej liečby bol takmer 15 rokov. Raritnou je pacientka aj vzhľadom na prítomnosť monoklonálnej gamapatie triedy IgG, pričom prítomný je dokonca zdvojený paraproteín IgG kappa. Druhá kazuistika dokumentuje pacientku so Schnitzlerovej syndrómom s typickými klinickými príznakmi, ale s diagnostickým omeškaním približne 10 rokov.
Opis kazuistického prípadu 1
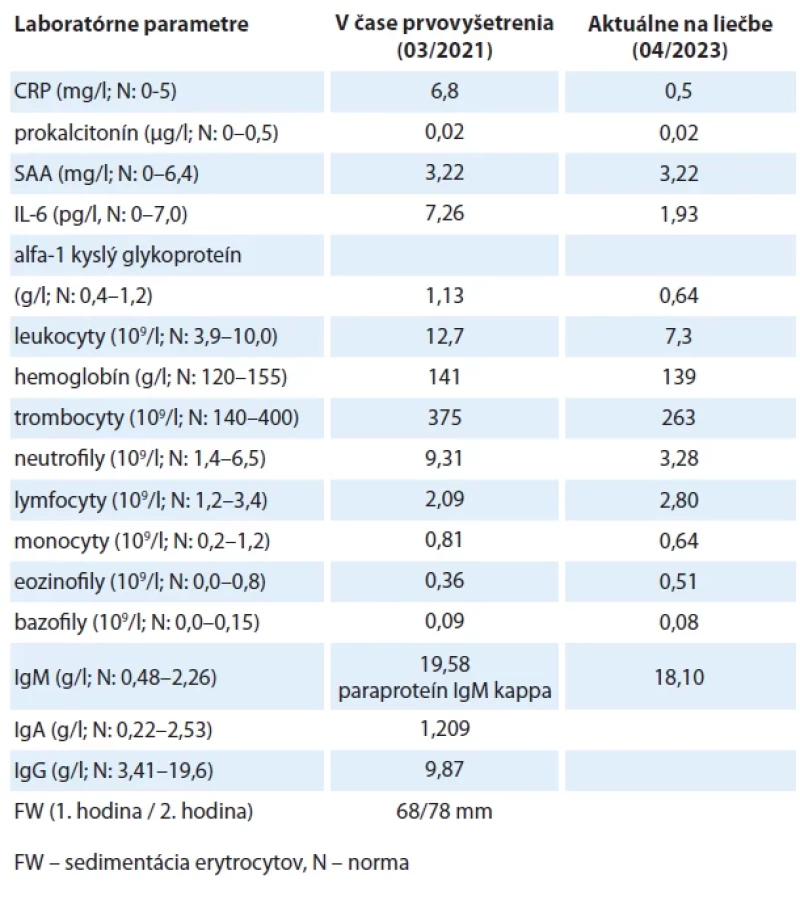
Pacientka bola vyšetrená na Ambulancii klinickej imunológie a alergológie Fakultnej nemocnice Trnava prvý krát v roku 2021 ako 37-ročná. Od prvého roku života bola sledovaná s diagnózou chronickej tubulointersticiálnej nefritídy, pričom prvýkrát bola vyšetrená hematológom v roku 2007 pre vysoké IgG, v krvnom obraze bola prítomná tiež trombocytóza. Hematológom bol stav hodnotený ako polyklonálna gamapatia a reaktívne zmeny v krvotvorbe. Pre pridružené bolesti kostí a kĺbov od roku 2016 bola vyšetrená v roku 2017 reumatológom, pričom stav bol hodnotený ako seropozitívna reumatoidná artritída. V laboratórnom obraze bola perzistujúca zvýšená zápalová aktivita – mierne zvýšený CRP, vysoký reumatoidný faktor (RF). Reumatológom bola nasadená liečba metrotrexátom a pre zhoršenie stavu aj systémové kortikoidy (40 mg prednison denne per os), ale bez efektu. Pre neuspokojivý stav bola pacientka vyšetrená opakovane v Národnom ústave reumatických chorôb, stav bol následne prehodnotený ako Sjögrenov syndróm vzhľadom na pozitívne autoprotilátky SS-A a SS-B, prítomná bola hypokomplementémia C3 a C4. Do liečby boli pridané antimalariká, ale stav bol bez zlepšenia či skôr s tendenciou k zhoršeniu. Pacientka bola prvýkrát vyšetrená na našej imunologickej ambulancii v roku 2021 s diagnózou chronickej žihľavky. Urtikária bola anamnesticky trvajúca od roku 2017 a objavila sa počas tehotenstva. Prejavy urtikárie boli denné, s výnimkou tváre celotelové, dominujúce najmä na dolných končatinách. Zaujímavými sú makroskopicky dve rozdielne typy žihľavky, na dolných končatinách až sýtočervenej a fialovej farby, imponujúce vaskulitického charakteru (obr. 1, 2), či klasickejšie urtikariálne prejavy dominujúce na ostatných častiach tela (obr. 3, 4). Pacientka udávala pocit skôr pálenia ako svrbenia. V klinickom obraze okrem žihľavky a artralgií boli prítomné aj recidivujúce bolesti brucha. V roku 2021 bola pacientka približne s mesačnou periodicitou, celkovo 7×, vyšetrovaná na ambulanciách urgentného príjmu so záverom akútnej gastroenteritídy. Gastrointestinálne ťažkosti vždy odozneli približne po 3 dňoch na symptomatickej liečbe. Laboratórne pretrvávala zvýšená zápalová aktivita – zvýšený CRP (10–29 mg/l; norma (N): 0–5), CRP vo fyziologickej hodnote sme u pacientky za posledných 5 rokov ani v dokumentácii nezaznamenali. V krvnom obraze prítomná opakovane tiež leukocytóza s neutrofíliou a trombocytózou. V biochemických parametroch ďalej prítomný vysoký sérový amyloid A (73,10 mg/l; N: 0–6,4), vysoký RF (81–295 IU/ml; N: 0–15), zníženie C3 (0,35–0,56 g/l; N: 0,9–1,8) a C4 (0,03–0,07 g/l; N: 0,1–0,4) komplementu, pozitívna autoprotilátková aktivita SS-A a SS-B. V paneli špecifických IgE bola zaznamenaná pozitivita pre alergény plesní (bez súvisu s klinickými ťažkosťami). Pre pridruženú generalizovanú lymfadenopatiu a fluidotorax bola v roku 2021 pacientka prijatá na diagnostickú hospitalizáciu na Kliniku vnútorného lekárstva FN Trnava. Počas hospitalizácie bola realizovaná biopsia lymfatickej uzliny, kde prítomná nešpecifická etiologicky nepríznačná lymfadenitída, v kostnej dreni bola atypická malobunková B-lymfoproliferácia nejasnej etiologie (do 10 %). S podozrením na autoinflamačný charakter ťažkostí bola následne pacientka odoslaná do Centra pre periodické horúčky (expertízne pracovisko Ministerstva zdravotníctva SR pre zriedkavé ochorenia) v Univerzitnej nemocnici v Martine. Tam v hematologickom náleze prítomný zdvojený paraproteín triedy IgG kappa. Realizované bolo genetické vyšetrenie na vrodené monogénové autoinflamačné ochorenia (panel vybraných génov) s negatívnym nálezom. V októbri 2022 bola pacientka akútne hospitalizovaná na Klinike vnútorného lekárstva FN Trnava pre zápalový syndróm s dyspnoe, urtikáriou, lymfadenopatiou a vysokými febrilitami. V laboratórnych parametroch vysoká elevácia CRP (319 mg/l; N: 0–5), IL-6 (55,8 pg/ml; N: 0–4,4) fibrinogénu (7,18 g/l; N: 1,8–3,5). Pre riziko z omeškania bola nasadená antibiotická empirická liečba, reumatológom bol stav hodnotený ako vzplanutie systémovej choroby spojiva – suspektný systémový lupus erythematosus, preto bola vzhľadom na akútny stav a nepriaznivý klinický obraz ordinovaná pulzná kortikoterapia a cyklofosfamid. Stav však naďalej progredoval, na RTG hrudníka bol prítomný obraz syndrómu akútnej dychovej tiesne (ARDS) (obr. 5) a stav bol vyhodnotený ako syndróm aktivovaných makrofágov v rámci overlapu autoimunity a autoinflamácie. Z tohto dôvodu bola podávaná 7 dní anakinra s výrazným zlepšením stavu, poklesom zápalovej aktivity a možnosťou demitácie pacientky. Stav sme po splnení Strassburgských kritérií uzatvorili ako Schnitzlerovej syndróm. Dočasne bola pacientke na skúšku vydaná anakinra, ktorú s dobrým efektom aplikovala systémom podľa potreby pri bolestiach brucha s hyperpyrexiami. Následne bola nastavená na liečbu kanakinumabom (v dávke 150 mg a neskôr 300 mg subkutánne raz za 4 týždne), pri ktorej bolo však pozorované len čiastočné zlepšenie stavu. V menšej miere pretrvávala urtikária a aj bolesti kĺbov a kostí, bez zmeny perzistovala elevácia zápalovej aktivity. Bolesti brucha pretrvávali cyklicky rekurentne mesačne a bolo nutné aj opakované podávanie anakinry. Vzhľadom na parciálny efekt kanakinumabu bola následne zrealizovaná zmena liečby na pravidelné podávanie anakinry s dobrým efektom. Aktuálne si pacientka dávkuje anakinru v dávke 100 mg subkutánne obdeň v domácom prostredí, pričom v prípade pocitu vzplanutia ochorenia si prechodne dávkovanie navyšuje na denné, raz za čas vyžaduje podávanie aj 2× denne. Pri tejto liečbe nemá takmer žiadne klinické ťažkosti. Laboratórne parametre dokumentujeme v tab. 2.






Opis kazuistického prípadu 2
Pacientka bola prvýkrát vyšetrená v Centre pre periodické horúčky v Univerzitnej nemocnici v Martine v roku 2021 vo veku 74 rokov. Pacientka bola v tom čase dispenzarizovaná pre viaceré chronické diagnózy (anamnesticky DM 1. typu na diéte a perorálnych antidiabetikách, chronická tubulointersticiálna nefritída / chronická choroba obličiek 1 podľa klasifikácie Kidney Disease: Improving Global Outcomes (KDIGO), vredová choroba gastroduodena, suspektný adenóm nadobličky l. sin. bez sekrečnej aktivity, polyp rekta a sigmy, chronická obstrukčná choroba pľúc v remisii, v minulosti bola vzhľadom na pozitívitu testu interferon-γ release assay (IGRA) preliečená antituberkulotikami). Rovnako roky bola dispenzarizovaná hematológom pre monoklonálnu gamapatiu nejasného významu (monoclonal gammopathy of undetermined significance – MGUS) a dermatológom pre chronickú idiopatickú urtikáriu nereagujúcu na liečbu antihistaminikami a kortikoidmi. U pacientky boli počas života realizované viaceré chirurgické výkony (stav po tonzilektómii, cholecystektómii, hysterektómii, appendektómii a operácii štítnej žľazy). Opakovane realizovaná kolonoskopia s negatívnym nálezom, pre lymfadenopatiu aj biopsia lymfatických uzlín s negatívnym nálezom. Vyšetrenie širokého panelu autoprotilátok bolo opakovane negatívne. V čase prvovyšetrenia pacientka prichádza pre 10 rokov trvajúce ťažkosti v zmysle chronickej urtikárie, exantému, bolestí kostí a kĺbov a nešpecifických celkových príznakov (akcentácia únavy počas febrilného ataku). Urtikária sa objavovala typicky bez pruritu, skôr pacientka opisovala pocit pálenia. V ostatnom klinickom obraze boli prítomné horúčky, hmotnostný úbytok, slabosť, únava, vertigo, artralgia a bolesti kostí. Pre nález monoklonálnej gamapatie IgM – elevácia voľných reťazcov kappa (28,14 mg/l; N: 3,30–19,40; index kappa/lambda 2,20) v kontexte celkových klinických príznakov boli splnené kritériá pre Schnitzlerovej syndróm. Pacientka bola následne nastavená na liečbu anakinrou v prvej línii v dennej aplikácii (100 mg s.c. à 24 hod.). Doplnili sme genetické vyšetrenie celého klinického exómu – bez nálezu patogénnej mutácie v asociácii so stavom a ťažkosťami pacientky. Laboratórny obraz vybraných parametrov pri prvom vyšetrení a ich zmeny počas liečby anakinrou dokumentujeme v tab. 3. U pacientky na liečbe anakinrou po treťom podaní takmer kompletne vymizli kožné klinické príznaky. Pri snahe o redukciu dávky (podávanie v režime 100 mg s.c. à 48 hodín) sme zaznamenali relaps klinických ťažkostí, preto pokračujeme v dennom podávaní. Pacientka sa má aktuálne klinicky veľmi dobre, liečbu toleruje bez lokálnych nežiadúcich účinkov, príznaky sú minimálne, laboratórny nález stabilný, zápalová aktivita je negatívna. Pacientka je pravidelne dispenzarizovaná aj hematológom, zatiaľ bez progresie do onkologického ochorenia. Komplikácie v zmysle rozvoja MAS sme u tejto pacientky nezaznamenali.
Diskusia
Autoinflamačné ochorenia sa stále radia medzi vzácne ochorenia, aj keď diagnostika vzhľadom na špecializované centrá a dostupnosť genetického vyšetrenia napreduje a počet pacientov aj pomenovaných nových ochorení pribúda. Autoinflamačné ochorenia môžeme rozdeliť na monogénové, u ktorých je známa a dokázateľná mutácia, a na ochorenia komplexné, kde táto mutácia známa nie je a predpokladá sa polygénová etiológia. Monogénové autoinflamácie sa diagnostikujú na základe genetického vyšetrenia s dokázaním defi - novanej mutácie, komplexné autoinflamácie podľa akceptovaných medzinárodných kritérií. Väčšina autoinflamácií sa manifestuje už v detskom veku, iba malý počet autoinflamačných ochorení sa manifestuje až v dospelosti. Typickým príkladom monogénového autoinflamačného ochorenia detského veku je HyperIgD syndróm a CAPS (cryopyrin-associated periodic syndrom), príkladom komplexného ochorenia detského veku je PFAPA (periodic fever with aphthous stomatitis, pharyngitis, and adenitis) syndróm. Z monogénových chorôb v dospelosti spomeňme VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory somatic syndrome) syndróm, z komplexných autoinflamácii v dospelosti Stillova choroba dospelých (adult onset Still‘s disease – AOSD)) a Schnitzlerovej syndróm. S variabilným výskytom ako v detstve, tak aj v dospelosti, je TRAPS alebo FMF [8]. Dve zo spomenutých ochorení s prvou manifestáciou v dospelosti sú reumatologickou literatúrou radené do skupiny autoinflamačných ochorení, hematologickou literatúrou do skupiny krvných ochorení, nakoľko sú pravidelne sprevádzané hematologickými laboratórnymi zmenami. Jedná sa v prvom rade o Schnitzlerovej syndróm, ktorý je hematologickou literatúrou zaradený medzi gamapatie, pretože je pravidelne sprevádzaný monoklonálnym imunoglobulínom. Druhou autoinflamačnou chorobou, ktorá patrí do skupiny hematologických ochorení typu myelodysplastického syndrómu, je VEXAS syndróm. Schnitzlerovej syndróm je raritné ochorenie s významným negatívnym vplyvom na kvalitu života. Do roku 2022 bolo celkovo na svete známych približne 300 pacientov [6]. Ochorenie je však v skutočnosti pravdepodobne významne poddiagnostikované. Už v práci z Mayo Clinic v USA autori uvádzajú, že v rokoch 1972–2010 mali diagnostikovaných iba 16 pacientov so Schnitzlerovej syndrómom. V sledovaní však mali 4 103 pacientov s monoklonálnou gamapatiou triedy IgM nejasného významu (MGUS-IgM) za účelom včasnej identifikácie prípadnej transformácie do Waldenströmovej makroglobulinémie. V tejto skupine pacientov vykonali skríning Schnitzlerovej syndrómu a odhalili ďalších 46 nerozpoznaných prípadov. Tento počet teda takmer trojnásobne prevyšoval počet diagnostikovaných prípadov. Išlo o pacientov, ktorý dlhodobejšie udávali najmä febrílie či subfebrílie (54 %) a/alebo bolesti kostí a kĺbov (78 %), k správnemu dodiagnostikovaniu to však hematológov neviedlo, nakoľko o vzácnom Schnitzlerovej syndróme pravdepodobne ani nevedeli. Autori tejto zatiaľ najväčšej analýzy prišli k záveru, že Schnitzlerovej syndróm môže byť vyjadrený až u 1,5 % pacientov s MGUS typu IgM či Waldenströmovou makroglobulinémiou [9]. My len dodávane, že sa nesmie zabúdať ani na pacientov pri Schnitzlerovej syndróme s raritnejšou monoklonálnou gamapatiou IgG. Problematická diagnostika s dlhým diagnostickým omeškaním od vzniku ťažkostí po správnu diagnózu a adekvátnu terapiu ohrozuje pacienta aj priamo na živote. Pacienti totiž bývajú s nejasnou, eventuálne nesprávnou reumatickou diagnózou, liečení rozličnými imunosupresívami bez podstatného efektu, navyše s rizikom aj závažných komplikácii navodených liečbou. My v našich kazuistikách opisujeme prípady dvoch žien, pričom v dostupných prácach pomer pacientov v kazuistikách je 1,6 : 1 v prospech mužov [10]. U našej pacientky v prvej kazuistike čas, ktorý uplynul od prvého hematologického vyšetrenia v roku 2007 s nálezom hyper - gamaglobulinémie triedy IgG do stanovenia diagnózy Schnitzlerovej syndrómu a nasadenie cielenej liečby (anakinra) s významným zlepšením stavu pacientky, trval 15 rokov. Pacientka nasadenú liečbu od reumatológa navyše zle tolerovala, stav sa jej pri niektorých liekoch dokonca zhoršoval (pri metotrexáte pre zvažovanú reumatoidnú artritídu bola pozorovaná progresia kĺbových aj gastrointestinálnych ťažkostí, pri kortikoidoch cushingoidný habitus a liečba bola bez klinického efektu). Ďalším zaujímavým faktom je relatívne mladý vek našej pacientky, keďže Schnitzlerovej syndróm sa častejšie manifestuje vo vyššom veku. Väčšina pacientov je diagnostikovaná okolo 50 roku života [10], pričom v literatúre nachádzame údaj o najmladšom pacientovi vo veku 27 rokov [11]. Diagnostickým problémom u našej pacientky bola aj od roku 2007 prítomná hypergamaglobulinémia IgG, pričom hematológom bol nález opakovane hodnotený ako polyklonálna gamapatia, na inom pracovisku sa však v roku 2023 potvrdila monoklonálna gamapatia IgG, prítomný bol dokonca zdvojený paraproteín triedy IgG kappa, čo pravdepodobne mohlo spôsobiť predchádzajúce nesprávne laboratórne závery. Navyše, polyklonálne gamapatie sprevádzajú rozličné chronické zápalové stavy, vrátane niektorých infekčných, autoinflamačných či klasických reumatických ochorení [12]. Monoklonálna gamapatia v triede IgG je navyše pri Schnitzlerovej syndróme raritná. Podľa recentnej štúdie 281 pacientov so Schnitzlerovej syndrómom je monoklonálny IgM prítomný u väčšiny pacientov (94 %), pričom ľahký reťazec kappa je prevažne zošikmený (85 %) a IgG tvorí len menšinu prípadov (6 %) [13]. Pacientka bola tiež častou návštevníčkou chirurgického a interného urgentného príjmu pre závažné akútne bolesti brucha, ktoré boli opakovane hodnotené ako akútna gastroenteritída, bez nálezu náhlej príhody brušnej. Nápadnou bola približne mesačná periodicita ťažkostí. Z dostupných prác abdominalgie u pacientov so Schnitzlerovej syndrómom neboli opísané často, ale môžu sa v klinickom obraze vyskytovať [14]. Pred nastavením na biologickú liečbu bola u pacientky dlhodobo nutná aj analgetická liečba pre pretrvávajúce až imobilizujúce bolesti kĺbov, kostí aj brucha, tiež infúzna liečba cestou domácej ošetrovateľskej starostlivosti. U pacientky bola tiež potrebná opakovane akútna hospitalizácia, v roku 2021 pre torpídne hnačky, lymfadenopatiu, v roku 2022 pre vysoké febrility, artralgie a dyspnoe, pričom stav vyústil až do závažnej hyperinflamácie s obrazom MAS a ARDS, či v roku 2023 pre febrility, zhoršenie kožného výsevu a artralgie. U pacientky sme prechodne indikovali liečbu kanakinumabom najskôr v dávkovaní 150 mg mesačne, následne pre nedostatočný efekt 300 mg mesačne, uvedená liečba ale viedla len k čiastočnej úprave klinického stavu pacientky, najmä ústupu chronickej žihľavky, ale pretrvávali limitujúce artralgie, pričom sa stupňovali najmä na obdobie, kedy už pravdepodobne doznieval účinok kanakinumabu, tj. najmä posledný týždeň pred ďalším podaním. Z tohto dôvodu, ako aj v kontexte priaznivej terapeutickej odpovede na epizodicky podávanú anakinru pred liečbou kanakinumabom, sme sa rozhodli pacientku nastaviť na anakinru s pravidelným podávaním. Na porovnanie s literatúrou, pacient so Schnitzlerovej syndró - mom liečený kanakinumabom v schéme 150 mg každých 8 týždňov dosiahol remisiu ochorenia do 24 hodín od podania, pričom ku koncu 8 týždňového intervalu sa objavovali prvé kožné lézie [15]. Iní pacienti liečení kanakinumabom boli v remisii do objavenia sa prvých príznakov priemerne 72 dní po poslednej dávke [16]. Od nastavenia biologickej liečby anakinrou a vysadenia kortikoidov naša pacientka nebola vyšetrená na urgentnom príjme, nebola hospitalizovaná, upravila sa chronická urtikária, minimalizovali sa bolesti kĺbov, svalov a kostí a odozneli periodické bolesti brucha. Pacientka si anakinru podáva s.c. v dávke 100 mg obdeň, pričom v prípade subjektívneho pociťovania zhoršenia ťažkosti (flare-up ochorenia) si navýši podávanie na denné, výnimočne vyžaduje podanie 2× denne. Diagnostický problém u pacientky spôsobujú aj vysoko pozitívne autoprotilátky anti-SS-A a anti-SS-B, ktoré bývajú pozitívne najmä pri systémových autoimunitných ochoreniach ako systémový lupus erythematosus a Sjögrenov syndróm. Ani u našej pacientky ale nie je vylúčený overlap autoimunity a autoinflamácie, čo by vysvetlilo závažný priebeh ochorenia. Pacientka však na klasickú liečbu autoimunity dobre nereaguje, bez efektu boli podávané kortikoidy, cyklofosfamid, metotrexát, hydrochlorochín, nesteroidné antiflogistiká. V rámci diferenciálnej diagnostiky pri uvedenom priebehu ochorenia treba zvažovať najmä AOSD, kryopyrinopatie (ale diagnóza prakticky vylúčená genetickým vyšetrením) a rôzne urtikariálne vaskulitídy, najmä hypokomplementový typ [17]. V druhej kazuistike rovnako pozorujeme významné diagnostické oneskorenie, správna diagnóza v tomto prípade bola stanovená po 10 rokoch. Aj u tejto pacientky podávanie biologickej liečby viedlo k ústupu klinických ťažkostí. Hematologickú malignitu sme zatiaľ u našich pacientiek nezaznamenali, niektoré menšie práce naznačujú zvýšené riziko vzniku malignity pri monoklonálnej gamapatii IgG [18].
Záver
Diagnostika Schnitzlerovej syndrómu je zložitá a vzhľadom na absenciu jednoznačného laboratórneho markeru, ktorý by diagnózu potvrdil, aj zdĺhavá. Navyše, o raritných ochoreniach je aj v lekárskej obci stále nízke povedomie. Ak diagnózu nepoznáte, nemôžete ju správne stanoviť. Ako uvádzame vyššie, aj na takom pracovisku ako je Mayo Clinic, bol Schnitzlerovej syndróm značne poddiagnostikovaný. Široká je aj diferenciálna diagnostika, kde je nutné vylúčiť ochorenia nielen zo spektra autoimunitných a autoinflamačných. Myslieť treba pri hyperinflamačných stavoch aj na poškodenia parainfekčné či paraneoplastické. Prístup k pacientovi je multiodborový, pričom dominantnú úlohu hrajú imunoalergológ a hematológ. Okrem monoklonálnej gamapatie sú totiž pacienti až v 20 % ohrození rozvojom hematologickej malignity. Pri chronickej žihľavke sú často pacienti odosielaní imunoalergológovi alebo dermatológovi. Aj naša pacientka z prvej kazuistiky nekonzumovala dlhé roky mlieko s presvedčením, že ide o alergiu na bielkovinu kravského mlieka. Najdôležitejším aspektom správnej diagnózy je následne začatie správnej liečby. Moderné biologiká ako anakinra či kanakinumab významne zlepšili kvalitu života ako aj prognózu pacientov s touto raritnou diagnózou a umožňujú dosiahnutie kontroly nad ochorením u väčšiny z nich.
Sources
1. McDermott MF, Aksentijevich I, Galon J et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97 (1): 133–144. doi: 10.1016/S0092-8674 (00) 80721-7.
2. Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949; 12 (2): 234–347.
3. Adam Z, Krejčí M, Pour L et al. Schnitzler syndrome – report on a fourteen-year course of the disease and an overview of information on the disease. Vnitr Lek 2008; 54 (12): 1140–1153.
4. Kacar M, Pathak S, Savic S. Hereditary systemic autoinflammatory diseases and Schnitzler’s syndrome. Rheumatology 2019; 58 (Suppl 6): vi31–vi43. doi 10.1093/rheumatology/kez448.
5. Varettoni M, Arcaini L, Zibellini S et al. Prevalence and clinical significance of the MYD88 (L265) somatic mutation in Waldestrom’s macroglobulinemia and related lymphoid neoplasms. Blood 2013; 121 (13): 2522–2528. doi: 10.1182/blood-2012-09-457101.
6. Cong-Qiu C. Schnitzler syndrome and Schnitzler-like syndromes. Chin Med J 2022; 135 (10): 1190–1202. doi: 10.1097/CM9.0000000000002015.
7. Braud A, Lipsker D. Schnitzler syndrome: insights into its pathogenesis, clinical manifestations, and current management. Biomolecules 2024; 14 (6): 646. doi: 10.3390/biom14060646.
8. Betrains A, Staels F, Schrijvers R et al. Systemic autoinflammatory disease in adults. Autoimmun Rev 2021; 20 (4): 102774. doi: 10.1016/j.autrev.2021. 102774.
9. Jain T, Offord CP, Kyle RA et al. Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica 2013; 98 (10): 1581–1585. doi: 10.3324/haematol.2013. 084830.
10. Eiling E, Schroder J, Gross W et al. The Schnitzler syndrome: chronic urticaria and monoclonal gammopathy – an autoinflammatory syndrome? J Dtsch Dermatol Ges 2008; 6 (8): 626–631. doi: 10.1111/j.1610-0387.2008. 06627.x.
11. Więsik-Szewczyk E, Felis-Giemza A, Dziuk M et al. Schnitzler syndrome in a 27-year-old man: diagnostic and therapeutic dilemma in adult auto-inflammatory syndromes a case report and literature review. Int J Gen Med 2020; 13 : 713–719. doi: 10.2147/IJGM.S265482.
12. Shawky RM, Gaboon NEA. Hereditary periodic fever syndrome. Egypt J Med Hum Genet 2011; 12 (2): 117–125. doi: 10.1016/j.ejmhg.2011.07.005.
13. de Koning HD. Schnitzler’s syndrome: lessons from 281 cases. Clin Transl Allergy 2014; 4 : 41. doi: 10.1186/ 2045-7022-4-41.
14. Bursztejn A, Imperiale A, Lipsker D. Aortitis: a new feature of Schnitzler syndrome. JAAD Case Rep 2017; 3 (5): 454–456. doi: 10.1016/j.jdcr.2017.06.016.
15. Vanderschueren S, Knockaert D. Canakinumab in Schnitzler syndrome. Semin Arthritis Rheum 2013; 42 (4): 413–416. doi: 10.1016/j.semarthrit.2012.06.003.
16. de Koning HD, Schalkwijk S, van der Ven-Jongekrijg J et al. Sustained efficacy of the monoclonal anti-interleukin-1 beta antibody canakinumab in a 9-month trial in Schnitzler’s syndrome. Ann Rheum Dis 2013; 72 (10): 1634–1638. doi: 10.1136/annrheumdis-2012-202192.
17. Simon A, Asli B, Braun-Falco M et al. Schnitzler’s syndrome: diagnosis, treatment, and follow-up. Allergy 2013; 68 (5): 562–568. doi: 10.1111/all.12129.
18. Bashir M, Bettendorf B, Hariman R. A rare but fascinating disorder: case collection of patients with Schnitzler syndrome. Case Rep Rheumatol 2018; 2018 : 7041576. doi: 10.1155/2018/7041576.
19. Dingli D, Camilleri MJ. Schnitzler syndrome: clinical features and histopathology. [online]. https: //www.dovepress.com/schnitzler-syndrome-clinical-features-and-histopathology-peer-reviewed-fulltext-article-PLMI.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2025 Issue 2
Most read in this issue
- Schnitzlerovej syndróm – kazuistická séria
- Multimodální prehabilitace v onkogynekologii – případová studie
- Léčba unicentrické Castlemanovy choroby rituximabem, bendamustinem a dexametazonem zmenšila objem neoperabilního expanzivního ložiska v horním mediastinu a umožnila jeho radikální odstranění
- Ivosidenib v léčbě metastatického cholangiocelulárního karcinomu – kazuistika