
Gillespieho syndróm
Authors:
Adriána Zelníková 1,2; Anna Maurská 1; Ivana Paučinová 3; Michal Štubňa 1
Authors‘ workplace:
Očné oddelenie, Fakultná nemocnicas poliklinikou v Žiline
1; 1. lékařská fakulta, Univerzita Karlova, Praha
2; Oddelenie lekárskej genetiky, Fakultná nemocnicas poliklinikou v Žiline
3
Published in:
Čes-slov Pediat 2025; 80 (6): 295-298.
Category:
Case Report
doi:
https://doi.org/10.55095/CSPediatrie2025/035
Overview
Zelníková A, Maurská A, Paučinová I, Štubňa M. Gillespieho syndróm
Gillespieho syndróm je veľmi vzácne genetické ochorenie charakterizované triádou symptómov: čiastočnou anirídou, neprogresívnou cerebelárnou ataxiou a psychomotorickým oneskorením vývoja. Výskyt tohto ochorenia je extrémne raritný, s prevalenciou menej ako 1 : 1 000 000 novorodencov. Syndróm je asociovaný s patogénnymi variantmi v géne ITPR1. Tento gén je zodpovedný za reguláciu vápnikových kanálov v bunkách. V našej práci chceme prezentovať prípad 5-ročného chlapca s diagnózou Gillespieho syndrómu.
5-ročný chlapček bol odoslaný na očné vyšetrenie z dôvodu nálezu „výrazných pupíl“. Prvé symptómy sa u nášho pacienta objavili vo veku štyroch mesiacov vo forme hypotonických stavov a trasenia. Neurologické vyšetrenia potvrdili oneskorenie vývoja, pričom bola pozorovaná ataktická chôdza. Pri očnom vyšetrení bol prítomný horizontálny nystagmus a čiastočná anirídia na oboch očiach s charakteristickým vrúbkovaným okrajom zrenice. Genetické vyšetrenie odhalilo de novo vzniknutú variantu c.7622T>C v heterozygotnom stave v géne ITPR1 asociovanú s ochorením Gillespieho syndróm.
Gillespieho syndróm je zriedkavé a neprogresívne genetické ochorenie, ktoré sa prejavuje najmä očnými a neurologickými abnormalitami. Diagnóza je potvrdená na základe klinického obrazu a genetického testovania. Liečba je podporná a zameraná na zmiernenie ťažkostí.
Klíčová slova:
Gillespieho syndróm – anirídia – cerebelárna ataxia – de novo
Úvod
Gillespieho syndróm (GS) (G11.0, ORPHA 1065, OMIM 206 700) je extrémne zriedkavé genetické ochorenie, ktoré sa vyznačuje kombináciou očných a neurologických abnormalít. Prvýkrát ho opísal Frederick D. Gillespie v roku 1965, keď uviedol prípad 22-ročnej ženy a jej 19-ročného brata, ktorí vykazovali triádu vrodenej anirídie (absencia časti alebo celej dúhovky), cerebelárnej ataxie a psychomotorického oneskorenia vývoja. Od prvého opísania ochorenia bolo v odbornej literatúre zdokumentovaných menej než 100 prípadov, pričom molekulárne potvrdenie diagnózy bolo dostupné len u 37 z nich (29 s dominantnými variantmi, 8 s recesívnou formou). Klinický obraz pozostáva z triády neprogresívnej cerebelárnej ataxie, čiastočnej anirídie a mierneho psychomotorického oneskorenia. Očné prejavy GS sa týkajú najmä dúhovky. Anirídia je vždy prítomná, pričom vo väčšine prípadov má okraj zrenice charakteristický vrúbkovaný vzhľad. Ochorenie môže byť sprevádzané ďalšími očnými nálezmi, akými sú hypoplázia fovey a optického nervu, hypopigmentácia sietnice a/alebo pigmentové zmeny makuly, ktoré vedú k zníženej zrakovej ostrosti. Okrem očných abnormalít GS zahŕňa aj neurologické deficity, najmä axiálny hypotonus, problémy s koordináciou, dysartriu, statickú a kinetickú ataxiu.(1-4)
Kazuistika
Kazuistika sa zaoberá prípadom 5-ročného chlapca, ktorý bol odoslaný na očné vyšetrenie pre nález abnormálneho vzhľadu pupíl. Dieťa pochádza z tretej gravidity a tretieho pôrodu, narodil sa v 38. týždni tehotenstva plánovaným cisárskym rezom z indikácie predchádzajúcej sekcie matky. Gravidita bola riziková, od 35. týždňa bola komplikovaná hroziacim predčasným pôrodom. Pôrodná hmotnosť bola 3280 gramov, dĺžka 52 cm, Apgar skóre 10/10. Popôrodná adaptácia prebehla bez komplikácií. V rodinnej anamnéze bola u jeho sestry zaznamenaná epilepsia, ktorá bola liečená do 4 rokov, v súčasnosti je sledovaná bez medikácie. Matka bola vyšetrená pre poruchy pamäti, EEG odhalilo epileptiformnú aktivitu, ale bez potreby farmakologickej liečby. Matkin brat mal v detstve diagnostikovanú epilepsiu. Otec pacienta je sledovaný pre vertigo a jeho otec zomrel vo veku 70 rokov na karcinóm nadobličiek. V rodokmeni nebol zistený výskyt monogénových ochorení, vrodených vývojových chýb, reprodukčných strát ani konsanguinity. Pacient bol od 4 mesiacov sledovaný pre stavy trasu pripomínajúce Moorov reflex, bol hypotonický a sledovaný pre možné záchvaty trasu (shuddering attacks), ktoré boli neskôr potvrdené. Jeho psychomotorický vývoj bol oneskorený a sledovaný neurológom. V siedmom mesiaci života mu bola pre asymetrickú brachycefáliu indikovaná remodelačná ortéza, ktorú nosil približne štyri mesiace. Okrem toho bol sledovaný nefrológom pre dilatáciu dutého systému obličiek II. – III. stupňa a kardiológom, kde bol nález bez vrodenej vývojovej chyby srdca, prítomná len stopová mitrálna insuficiencia. Pacient bol taktiež sledovaný pre intelektový deficit mierneho stupňa a pre poruchy reči navštevoval logopéda. V druhom roku života sa pri MR CNS zistila solitárna lézia v spleniu corpus callosum a punktiformná nešpecifická lézia v bielej hmote vpravo, ktoré boli stacionárne. Kontrolné vyšetrenia neodhalili progresiu. Metabolický skríning, počas ktorého bol testovaný aj na Pompeho chorobu a iné dedičné metabolické poruchy, bol negatívny. Jeho psychomotorický vývoj pokračoval s oneskorením, pričom pacient začal samostatne chodiť vo veku 2 rokov a 9 mesiacov, ale chôdza bola ataktická a zhoršovala sa pri infekčných ochoreniach alebo strese. Pri neurologických kontrolách bola prítomná ataxia a známky polyneuropatie. Na genetickom vyšetrení bol karyotyp pacienta 46,XY, teda normálny mužský karyotyp bez štrukturálnych alebo numerických aberácií. ArrayCGH
neodhalila žiadne chromozomové imbalancie. Analýza génu FXN metódou TP-PCR a fluorescenčnou PCR s fragmentačnou analýzou nepotvrdila expanziu GAA traktu v géne FXN. Pri vyšetrení spinocerebelárnej ataxie typov 1, 2, 3, 6 a 7 (gény ATXN1, ATXN2, ATXN3, CACNA1A a ATXN7) fragmentačnou analýzou neboli detegované expandované alely, t. j. SCA 1, 2, 3, 6 a 7 sa nepotvrdili. Masívne paralelné sekvenovanie panelu 240 génov asociovaných s ataxiami odhalilo u pacienta heterozygotný missense variant c.7622T>C p.(Leu2541Pro) v géne ITPR1. Zistený variant nebol doteraz popísaný v žiadnej literatúre ani v databázach patogénnych či populačných variantov; napriek tomu je lokalizovaný v oblasti génu s vysokým výskytom známych kauzálnych mutácií a viaceré in silico predikčné nástroje (PolyPhen-2, SIFT, MutationTaster) naznačujú jeho škodlivosť. Podľa kritérií ACMG teda spĺňa PM1 (lokalizácia v kritickej doméne), PM2 (absencia v kontrolnej populácii) a PP3 (podpora patogenity in silico), a bol pôvodne zaradený ako variant nejasného klinického významu. Asi rok po našej prvej diagnostike databáza Varsome tento nález preklasifikovala na „pravdepodobne patogénny“ a spätne ho aj program Franklin označil stupňom 4/5 (pravdepodobne patogénny). Patogénne varianty v géne ITPR1 sú známe asociovať so spinocerebelárnou ataxiou typu 15, spinocerebelárnou ataxiou typu 29 a s Gillespieho syndrómom. Aby sme overili, či je tento nález u dieťaťa de novo, vykonali sme segregačnú analýzu u rodičov: variant nebol prítomný ani u jedného z nich, čo potvrdilo de novo vznik. Pacient absolvoval očné vyšetrenie vzhľadom na suspektný nález na zreniciach. Pri oftalmologickom vyšetrení bol zistený bilaterálny nález zodpovedajúci opísaným abnormalitám. Očné vyšetrenie ukázalo fixované rozšírené zrenice nereagujúce na svetlo ani na pilokarpín, horizontálny nystagmus a čiastočnú anirídiu na oboch očiach s vrúbkovaným okrajom zrenice (obr. 1). Vyšetrenie očného pozadia nepreukázalo patologický nález. Klinické príznaky u dieťaťa sú v zhode s popisom Gillespieho syndrómu (ataxia, intelektuálny deficit, hypotónia a hypoplázia dúhovky). Na základe nálezu de novo variantu a fentypovej zhody sme kazuistiku uzavreli ako Gillespieho syndróm. Gillespieho syndróm sa podľa literatúry môže dediť autozómovo dominantne aj autozómovo recesívne, v závislosti od konkrétneho genetického pozadia. V našom prípade ide o de novo vzniknutý variant v géne ITPR1, čo potvrdzuje neprítomnosť daného variantu u oboch rodičov, a preto riziko rekurencie u súrodencov je nízke (obr. 2).(5,6)
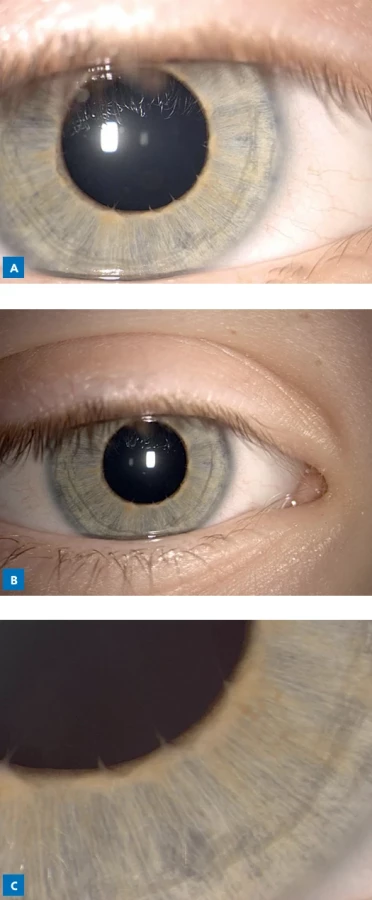

Diskusia
Gillespieho syndróm je extrémne zriedkavé ochorenie (prevalencia < 1 : 1 000 000), ktorého dedičnosť môže byť autozómovo dominantná aj autozómovo recesívna, pričom väčšina dominantných variantov vzniká de novo. Klinické príznaky sa objavujú už v prvom roku života a zahŕňajú fixované dilatované zrenice a vrodenú hypotóniu. Typickým očným nálezom je hypoplázia dúhovky s vrúbkovaným pupilárnym okrajom, z ktorého vlákna dúhovky pravidelne vystupujú na prednú plochu šošovky. Medzi extraokulárne prejavy patrí neprogresívna cerebelárna hypoplázia vedúca k ataxii, variabilný kognitívny deficit (spravidla mierny) a niekedy mierna faciálna dysmorfia, vyznačujúca sa vysokým čelom, hypertelorizmom, epikantami, anteverziou nozdrí a úzkou hornou perou. Existuje prípadová štúdia publikovaná v časopise Cureus, ktorá opisuje pacienta s Gillespieho syndrómom, ktorý mal bilaterálnu aniridiu a ataxiu, ale bez mentálneho postihnutia a bez známej rodinnej anamnézy. Táto štúdia naznačuje, že mentálne postihnutie nemusí byť nevyhnutnou klinickou črtou.(7)
Gillespieho syndróm je spôsobený buď monoalelickými, alebo bialelickými patogénnymi variantmi v géne ITPR1 (chromozóm 3p26.1); hlásené sú missense, nonsense, frameshift aj splice-site mutácie. Gén ITPR1 kóduje receptor inozitol-1,4,5-trisfosfátu typu 1, intracelulárny transmembránový kanál, ktorý uvoľňuje vápnik z endoplazmatického retikula v reakcii na IP₃. Proteínový komplex ITPR1 je homotetramér, pričom každá podjednotka obsahuje cytoplazmatické väzbové miesto pre IP₃ a krátku C-terminálnu oblasť, šesť transmembránových domén tvoriacich pór kanála a intertransmembránové slučky v lumene ER. Niektoré prípady podobného Gillespiemu sú spojené aj s variantmi v géne PAX6, avšak primárne ide o odchýlky v ITPR1. Ak je prítomný dominantný patogénny variant ITPR1, riziko prenosu na potomstvo je 50 % bez ohľadu na pohlavie. V prípade, že rodičia nie sú nosičmi rovnakého variantu, ide o de novo mutáciu, a hoci je riziko pre ďalšie dieťa daných rodičov nízke, nemožno vylúčiť možnosť germinálnej mozaicity. Keďže postihnuté dieťa má 50 % šancu odovzdať ochorenie svojim deťom, je dostupná prenatálna aj preimplantačná genetická diagnostika.(5,7–9)
Na rozdiel od Gillespieho syndrómu je klasická anirídia najčastejšie spôsobená heterozygotnými stratofunkčnými mutáciami v géne PAX6 a býva spojená s hypopláziou fovey, ktorá predstavuje charakteristický oftalmologický znak.(1) Klinicky sa symptómy anirídie delia do dvoch hlavných kategórií: vrodené a získané, pričom obe ovplyvňujú zrak. Vrodené abnormality, ako sú hypoplázia zrakového nervu a hypoplázia fovey, ovplyvňujú zrak už pri narodení. Tieto abnormality sú často spojené s vrodeným nystagmom, ktorý naznačuje skoré a často trvalé zrakové postihnutie. S pribúdajúcim vekom sa objavujú aj ďalšie progresívne zmeny. Vrodený deficit kmeňových buniek limbu vedie k postupnému poškodeniu rohovky, ktoré sa prejavuje hustou opacifikáciou (pozorovanými až u 80 % pacientov). Šedý zákal postihuje 40–80 % pacientov, pričom zmeny na šošovke sa môžu prejaviť už v detstve, no významne ovplyvnia zrak až v dospelom veku. Vážny vplyv na zrak ma glaukóm a jeho poškodenie zrakového nervu. Aniridický glaukóm je kombinovaný glaukóm s otvoreným a uzavretým uhlom, ktorý býva často rezistentný na liečbu.(2)
Anirídia sa však môže vyskytovať aj ako súčasť iných genetických syndrómov, napríklad WAGR syndrómu, ktorý je podmienený deléciou oblasti 11p13 zahŕňajúcou gény PAX6 a WT1. Ide o zriedkavé genetické ochorenie charakterizované prítomnosťou Wilmsovho tumoru, anirídie, genitourinárnych abnormalít a rôzneho stupňa vývinového oneskorenia. Okrem týchto klasických znakov sa u postihnutých jedincov môže rozvinúť aj obezita a renálne zlyhávanie. Anirídia sa môže objaviť aj pri iných syndrómoch, ako je syndróm dysfunkcie hladkých svalov spojený s mutáciami v géne ACTA2 alebo pri syndrómoch spojených s génmi FOXC1, PITX2 a BDNF.(4) Pri diferenciálnej diagnostike je preto nevyhnutné zohľadniť celkový fenotyp pacienta a využiť genetické testovanie na vylúčenie iných možných príčin. V prezentovanom prípade boli alternatívne diagnózy vylúčené na základe klinického obrazu a výsledkov genetických analýz.(1)
Záver
Gillespieho syndróm patrí medzi extrémne zriedkavé genetické ochorenia. Jeho diagnostika si vyžaduje multidisciplinárny prístup a absolvovanie genetického vyšetrenia, ako bolo aj v prípade nášho pacienta. Klinické podozrenie by sa malo zvážiť u každého hypotónneho dieťaťa s fixovanými, dilatovanými zrenicami. Čiastočná anirídia je kľúčovým diagnostickým znakom, prítomným zatiaľ u všetkých popisovaných prípadov. V článku poukazujeme na potrebu poznania aj takto zriedkavej patológie, aby sa včasne rozpoznali symptómy a bola nastavená podporná liečba, ktorá môže zlepšiť kvalitu života pacientov. Nakoľko približne 5 % prípadov klasickej anirídie zostáva nevysvetlených, môžu byť tieto prípady spôsobené novými mechanizmami narúšajúcimi známe gény alebo mutáciami na nových lokusoch, ktoré môžeme objasniť metódou sekvenovania celého genómu (whole genome sequencing).(1)
Sources
1. Hall HN, Williamson KA, FitzPatrick DR. The genetic architecture of aniridia and Gillespie syndrome. Hum Genet 2019; 138(8-9): 881–898.
2. Balikov, DA, Jacobson A, Prasov L. Glaucoma syndromes: insights into glaucoma genetics and pathogenesis from monogenic syndromic disorders. Genes 2021; 12(9): 1403.
3. Ciaccio C, Taddei M, Pantaleoni C, et al. Phenotypic spectrum and natural history of Gillespie syndrome. An updated literature review with 2 new cases. Cerebellum 2024.
4. Tripathy K, Salini B. Aniridia. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing 2024.
5. Gerber S, Alzayady KJ, Burglen L, et al. Recessive and dominant de novo ITPR1 mutations cause Gillespie syndrome. Am J Hum Genet 2016; 98(5): 971–980.
6. Keehan L, Jiang MM, Li X, et al.; Undiagnosed Diseases Network. A novel de novo intronic variant in ITPR1 causes Gillespie syndrome. Am J Med Genet A 2021; 185(8): 2315–2324.
7. Singh G, Narahari S. A case of Gillespie syndrome with atypical presentation. Cureus 2022; 14(11): e31341.
8. Nabih O, Hamdani H, ELMaaloum L, et al. Gillespie syndrome: An atypical form and review of the literature. Ann Med Surg (Lond) 2022; 74 : 103244.
9. Stendel C, Wagner M, Rudolph G, Klopstock T. Gillespie’s syndrome with minor cerebellar involvement and no intellectual disability associated with a novel ITPR1 mutation: report of a case and literature review. Neuropediatrics 2019; 50(6): 382–386.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2025 Issue 6
Most read in this issue
- Nonakcidentální intoxikace u českých adolescentů od roku 2011 do roku 2024: násobný nárůst četnosti, proměny užitých látek a vliv pandemie covidu-19
- Jak v prvních dvou letech života rostou předčasně narozené české děti a jak jejich růst kvalitně hodnotit?
- Diferenciální diagnostika poruch vědomí u dětí
- Nové trendy v manažmente anafylaxie u detských pacientov