
Rozsah reportování při vyšetření predispozice k nejčastějším solidním nádorům dospělého věku pomocí sekvenování nové generace
Authors:
J. Soukupová 1; M. Koudová 2; L. Foretová 3; V. Krutílková 4; V. Curtisová 5; I. Blaháková 6; I. Dolinová 7; P. Dušková 8; L. Faldynová 9; M. Fišer 10; N. Friedová 11,12; M. Gančarčíková 13; Š. Chvojka 2; M. Janatová 1; M. Janíková 5; P. Kleiblová 1,12; K. Kyselová 14; E. Macháčková 3; R. Měch 15; R. Michalovská 16; M. Nečesánková 17; Z. Spurná 10; M. Šedinová 18; M. Šenkeříková 19; Z. Šimková 20; J. Šimová 21; I. Šubrt 22; S. Tavandzis 4; P. Tesner 23; J. Turňová 24; Z. Vlčková 16; M. Vočka 12,25; Konsorcium Czecanca 1; N/A pracovní skupina Onkogenetika SLG ČLS JEP 1; Z. Kleibl 1
Authors‘ workplace:
Ústav lékařské biochemie a laboratorní diagnostiky 1. LF UK a VFN v Praze
1; GENNET a GNTlabs by GENNET, Praha
2; Oddělení epidemiologie a genetiky nádorů, MOÚ Brno
3; Oddělení lékařské genetiky, Laboratoře AGEL a. s., Nový Jičín
4; Ústav lékařské genetiky FN Olomouc
5; Centrální laboratoř genomika, CEITEC MU, Brno
6; Oddělení genetiky a molekulární diagnostiky, Krajská nemocnice Liberec
7; Laboratoř molekulární biologie a genetiky, Nemocnice České Budějovice, a. s.
8; Oddělení lékařské genetiky, Ústav molekulární patologie a lékařské genetiky, FN Ostrava
9; Lékařská genetika, Prenet, Pardubice
10; Oddělení lékařské genetiky, FTN Praha
11; Ústav biologie a lékařské genetiky 1. LF UK a VFN v Praze
12; Ústav klinické biochemie a diagnostiky LF v Hradci Králové UK a FN Hradec Králové
13; Centrum lékařské genetiky, SPADIA LAB, a. s.
14; Fertimed, s. r. o., Olomouc
15; Oddělení klinické genetiky, GHC Genetics, Praha
16; Genetika Plzeň, s. r. o., Plzeň
17; Nemocnice Jihlava, p. o., Jihlava
18; Oddělení lékařské genetiky, FN Hradec Králové
19; Ambulance lékařské genetiky, Nemocnice České Budějovice, a. s.
20; EUC Laboratoře CGB a. s.
21; Ústav lékařské genetiky LF v Plzni UK a FN Plzeň
22; Ústav biologie a lékařské genetiky 2. LF UK a FN Motol a Homolka
23; Oddělení lékařské genetiky, Pronatal, Praha
24; Onkologická klinika 1. LF UK a VFN v Praze
25
Published in:
Klin Onkol 2026; 39(1): 56-63
Category:
Original Articles
doi:
https://doi.org/10.48095/ccko202656
Overview
Východiska: Genetické vyšetření dědičné predispozice k solidním nádorům pomocí sekvenování nové generace umožňuje analýzu širokého spektra genů a významně zvyšuje diagnostický výtěžek. Se zvyšujícím se rozsahem analyzovaných oblastí však roste pravděpodobnost záchytu tzv. sekundárních nálezů, tedy patogenních nebo pravděpodobně patogenních variant v genech, které nesouvisejí s primární indikací k vyšetření. Tyto nálezy přinášejí řadu praktických, klinických a etických otázek. Materiál a metody: V ČR vznikla v rámci konsorcia CZECANCA a pracovní skupiny Onkogenetika Společnosti lékařské genetiky a genomiky ČLS JEP iniciativa s cílem harmonizovat rozsah reportování při vyšetření nádorové predispozice v rozsahu panelu CZECANCA. Výsledky: Výsledkem konsenzu je doporučený rozsah variant a genů, které budou při vyšetření nádorové predispozice rutinně reportovány laboratořemi i bez výslovného požadavku indikujícího lékařského genetika. Závěr: Navržený harmonizovaný přístup k reportování sekundárních nálezů představuje praktický nástroj pro sjednocení laboratorní praxe v ČR. Dokument reflektuje současný stav poznání v oblasti onkogenetiky a bude pravidelně aktualizován v návaznosti na nové vědecké poznatky.
Klíčová slova:
genetické testování – geny – dědičné nádorové syndromy – konsenzus – vysoce účinné nukleotidové sekvenování
Úvod
Testování hereditární nádorové predispozice k solidním nádorům typicky dospělého věku prošlo v posledních letech významnou transformací. Současným standardem je panelové sekvenování různého rozsahu, které poskytuje vyšší diagnostický výtěžek a představuje výhodnější technologické řešení z hlediska finančního i časového. Vyšetřovány jsou typicky desítky genů až celý exom (whole exome sequencing – WES).
Zachycené varianty ve vyšetřovaných oblastech genomu jsou prioritizovány a následně v první úrovni klasifikovány na základě pravděpodobnosti patogenity do pěti tříd: 1) patogenní; 2) pravděpodobně patogenní; 3) varianty nejasného významu (variants of uncertain significance – VUS); 4) pravděpodobně benigní; 5) benigní. Za klinicky relevantní jsou považovány varianty klasifikované jako pravděpodobně patogenní či patogenní (P/LP), u nichž pravděpodobnost patogenity přesahuje 90 %. U klasifikovaných variant je následně ve druhé úrovni, které probíhá rovněž v laboratořích, zvažován vztah nalezených variant k fenotypu probanda. Za klinicky relevantní jsou považovány geny se střední až vysokou penetrancí (poměr šancí (OR) ≥ 2). Ve třetí úrovni hodnocení variant provádí lékařský genetik klinickou interpretaci genetického nálezu, v rámci které jsou zpřesňována rizika asociovaná s identifikovaným genotypem v kontextu osobní a rodinné anamnézy probanda, a dle výše absolutních rizik je doporučena odpovídající péče o nosiče/nosičky variant a jejich rodiny [1].
Mimo primární nálezy, které jsou spojeny s fenotypem testovaného probanda, nás větší rozsah testovaných oblastí staví před problém neočekávaných nálezů, jež nejsou spojeny s primárním důvodem testace, avšak mohou být pro testovaného a jeho rodinu klinicky relevantní. Neočekávané nálezy někdy bývají rozdělovány na nálezy sekundární a incidentální. Sekundární nálezy při vyšetření nádorové predispozice jsou spojeny s predispozicí k jinému nádorovému fenotypu, incidentální nálezy jsou spojeny s jinými, nenádorovými diagnózami. V posledních letech je však pro neočekávané nálezy preferován jednotný termín sekundární nálezy.
Vyšetření nádorové predispozice ve světě a v ČR
Problematika sekundárních nálezů přináší řadu praktických, etických a klinických dilemat. Rozsah vyšetření a reportování související s problematikou sekundárních nálezů je ve světě řešen různě v závislosti na zdravotním systému a organizaci genetického testování dědičné nádorové predispozice.
Na jedné straně narůstá pro nejčastější diagnózy, typicky karcinom prsu a ovaria, socioekonomicky výhodné „mainstream“ vyšetření malého počtu genů u všech nemocných [2]. Ve specifické izraelské populaci s vysokou prevalencí P/LP variant ve vysoce penetrantních genech BRCA1 a BRCA2 je dokonce pro zdravé ženy dostupný populační screening founder mutací v těchto genech [3]. Nicméně ve světě stále převládá testace více genů u podskupiny pacientů splňujících indikační kritéria, u kterých je pravděpodobnost záchytu P/LP variant vyšší, přičemž s rozvolňujícími se kritérii se rozšiřuje skupina testovaných pacientů. Nejčastěji jsou využívány panely genů specifické pro jednotlivé diagnózy, jejichž rozsah se liší mezi státy i mezi poskytovateli v rámci jednoho státu [2], a v závislosti na rostoucích znalostech se rozsah vyšetřovaných genů v čase dynamicky mění. V rámci racionalizace jsou někdy používány tzv. virtuální panely, což znamená, že bioinformaticky je hodnocena jen část reálně sekvenovaných genů, která je relevantní pro určitou nádorovou diagnózu [4]. Tyto přístupy eliminují sekundární nálezy, avšak v době testace nepodávají komplexní obraz o možné nádorové predispozici, která je komplikována typicky neúplnou penetrancí, případně variabilní expresivitou.
V ČR jsou používány velké multicancer panely až WES. Důvodem jsou racionalizace testování, které je decentralizováno mezi mnoho laboratoří s různým objemem testace, a způsob úhrady. V současné době plátci hradí vyšetření hereditární nádorové predispozice jedincům splňujícím indikační kritéria pro genetické testování 1× za život na konkrétní nádorovou diagnózu (dle klasifikace MKN-10 diagnózy C00-C97, Z80, Z85). Minimální požadovaný rozsah vyšetření aktuálně zahrnuje 22 genů (ATM, APC, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD50, RAD51C, RAD51D, STK11, TP53).
Existující doporučení pro sekundární nálezy
S rozšiřující se dostupností WES prováděného z řady indikací se objevily snahy o konsenzuální reportování, které vedly American College of Medical Genetics (ACMG) k publikování seznamu genů, jejichž neočekávané zachycené P/LP varianty (všechny či pouze definované varianty, u autozomálně recesivních (AR) onemocnění jen v případě bialelických P/LP variant) by měly být reportovány vždy, bez ohledu na primární důvod testování, pokud si pacient přeje být o takovém nálezu informován. V současné době (ACMG SF v3.3.; [5]) se jedná o 84 genů (box 1), které zahrnují i vysoce penetrantní nádorové predispoziční geny, dále např. geny spojené s maligními arytmiemi či geny zodpovědné za maligní hypertermii vyvolanou některými anestetiky. Seznam genů je pravidelně aktualizován a je odbornou komunitou široce přijímán [6].

Z pohledu prevence závažných AR onemocnění v populaci existují doporučení pro reportování přenašečství P/LP variant pro carrier testy, dle kterých je doporučeno reportovat P/LP varianty v genech spojených s AR onemocněním v případě frekvence heterozygotů v populaci převyšující 1/200 [7]. Při této populační frekvenci je riziko AR onemocnění u potomka > 1/800. Problémem je v případě některých genů velmi rozdílná frekvence P/LP variant v jednotlivých populacích [8]. V mezinárodní databázi GnomAD je zastoupení slovanské populace mizivé a pro určení frekvence heterozygotů v naší populaci je její využití pro řadu genů omezené.
Tvorba konsenzu
V ČR bylo vyšetření nádorové predispozice v roce 2024 provedeno u přibližně 12 000 jedinců a počet vyšetření meziročně narůstá. Klinicky relevantní nádorové predispoziční geny pro jednotlivé nádorové diagnózy nejsou jednoznačně definovány. Rozsah reportování se mezi laboratořemi liší, a vytváří tak velmi nepřehledné prostředí jak pro indikující a pečující lékaře, tak pro testované osoby.
Na jedné straně existuje snaha některých pracovišť reportovat všechny nálezy VUS/LP/P variant v celém rozsahu vyšetřovaných genů. Primárním důvodem je, s ohledem na absenci doporučení, zajištění právní ochrany v případě forenzního sporu. Dalším relevantním argumentem může být skutečnost, že fenotyp probanda asociovaný s nalezenou genetickou variantou v určitém nádorovém predispozičním genu nemusí být jednoznačně vyjádřen, a proto na něj při indikaci genetického vyšetření nebylo původně pomýšleno. Tento přístup „overreportování“ může vést k nadhodnocování asociovaných rizik a k nepřiměřeným preventivním opatřením, která ve svém důsledku představují minimálně psychickou zátěž pro pacienta a jeho rodinu, ale mohou vést k neadekvátním a nevratným preventivním výkonům a v neposlední řadě představují zbytečnou finanční zátěž pro zdravotní systém s omezenou kapacitou péče o rizikové jedince.
V rámci CZECANCA konsorcia a pracovní skupiny Onkogenetika SLG ČLS JEP vznikla v roce 2022 iniciativa definovat, které varianty a v jakých genech budou rutinně reportovány laboratoří směrem ke lékařskému genetikovi nad rámec požadovaných genů. Cílem je harmonizovat přístup k reportování výsledků genetických vyšetření a jejich následné interpretaci. Problematika rozsahu reportovaných variant byla pravidelně diskutována na společných setkáních konsorcia CZECANCA a pracovní skupiny Onkogenetika od roku 2022. Na 12. setkání (16. 5. 2024) jsme došli ke konsenzu, který shrnuje box 2.

V roce 2022 byla rovněž zahájena komplikovanější diskuze týkající se problematiky rozsahu reportovaných genů. Konsenzus byl tvořen online, a sice v rozsahu genů CZECANCA panelu v1.22 pro 226 genů [9]. Od června do listopadu 2024 se v iniciální fázi vyjádřili pracovníci ze 14 pracovišť (tab. 1) ke vhodnosti a důvodům rutinního reportování P/LP variant zachycených jako sekundární nálezy v uvedených genech. V případě AR dědičnosti bylo reportování monoalelických a bialelických variant diskutováno zvlášť. Výsledkem byla shoda na rutinním reportování P/LP variant v 54 genech, na nereportování ve 115 genech a u 59 genů nebyla shoda nalezena (schéma 1).
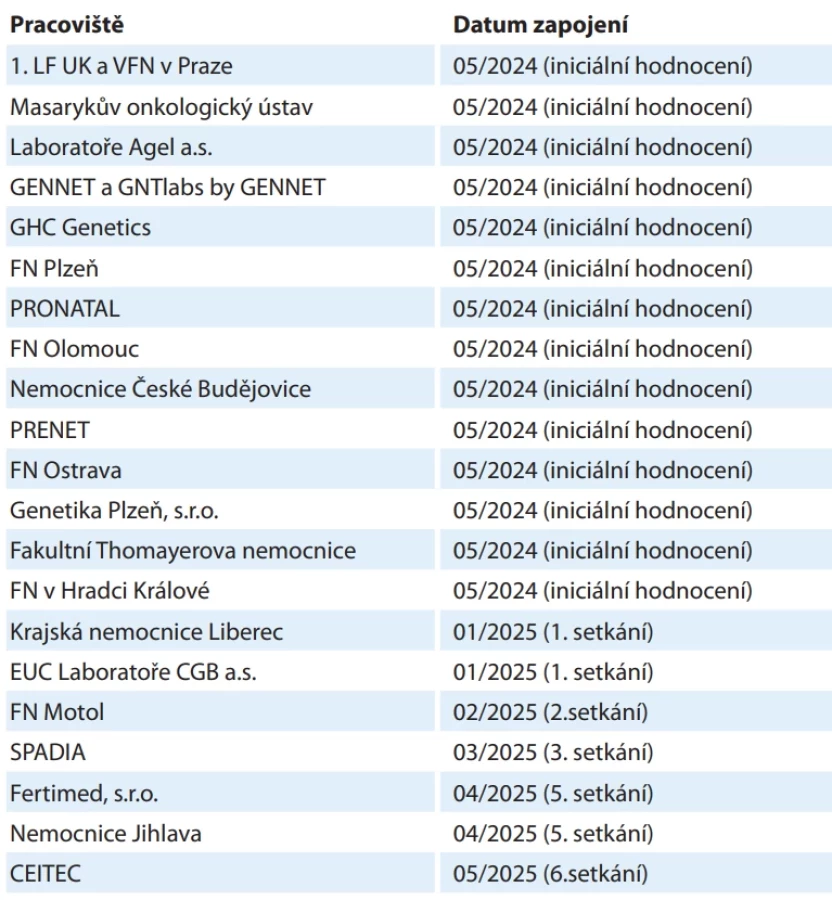

Následně od ledna do května 2025 bylo reportování P/LP variant v 59 sporných genech jednotlivě diskutováno v průběhu šesti online 90minutových setkání. Schůzek se zpočátku účastnilo 14 pracovišť, počet zapojených pracovišť postupně narostl až na 21 (tab. 1). Zvažována byla především míra penetrance a způsob jejího stanovení (zda data vychází z case-control analýz či z vysoce rizikových rodin se specifickým fenotypem), morbidita a mortalita asociovaného fenotypu, dostupnost a závažnost preventivních opatření či technické aspekty vlastního sekvenování (např. přítomnost pseudogenů) [10]. Diskutována byla rovněž specifika jednotlivých genů (typ dědičnosti, typy variant spojených s nádorovou predispozicí, případný imprinting apod.).
Výsledkem online setkání bylo stanovení konsenzu na rutinní reportování (tj. bez vyžádání indikujícím lékařem) sekundárně zachycených P/LP variant v 77 genech a nereportování ve 152 genech z laboratoře směrem k indikujícímu lékařskému genetikovi (tab. 2), který provádí následnou interpretaci nálezu v kontextu osobní a rodinné anamnézy. Tento konsenzuální rozsah reportování se týká vyšetření predispozice k nádorovým onemocněním primárně dospělého věku. V případě potřeby vyšetření některého ze 152 rutinně nereportovaných genů musí indikující lékařský genetik požadavek vyšetření tohoto genu jmenovitě uvést na žádance.
![Konsenzus týkající se rutinního reportování sekundárních nálezů P/LP variant při vyšetření nádorové predispozice (hodnoceny geny v rozsahu panelu CZECANCA v1.22) [9].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/da8a8c647f8f51701411249a497c4a83.png)
![pokračování. Konsenzus týkající se rutinního reportování sekundárních nálezů P/LP variant při vyšetření nádorové predispozice (hodnoceny geny v rozsahu panelu CZECANCA v1.22) [9].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/4304a79921daf4df85a53ee20559511c.png)
![pokračování. Konsenzus týkající se rutinního reportování sekundárních nálezů P/LP variant při vyšetření nádorové predispozice (hodnoceny geny v rozsahu panelu CZECANCA v1.22) [9].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/f0966b31f23dfa705be882052338189b.png)
V případě reportování extrémně vzácných P/LP variant spojených s mimořádně vzácnými syndromy (např. CDC73, PRKAR1A, PTCH1, SUFU apod.), které jsou asociovány mimo jiné s nádorovou manifestací a tradovanou vysokou penetrancí stanovenou na velmi omezeném počtu velmi vysoce rizikových rodin, panuje shoda na nutnosti postupovat při interpretaci nálezu mimořádně opatrně a ideálně nález individuálně diskutovat v rámci odborné komunity, protože penetrance těchto variant je spojena se značnou mírou nejistoty. Neexistuje navíc dostatek dat o penetranci těchto variant zachycených jako sekundární nálezy a hrozí zde vysoké riziko neadekvátních preventivních opatření jak u probanda, tak u jeho pozitivně testovaných, dosud onkologicky zdravých příbuzných. Lze předpokládat, že penetrance variant zachycených jako sekundární nálezy bude nižší. Tento fenomén je popsán u řady genů s dostatkem dat [11]. V některých případech (např. SDHA) již dokonce byla navržena rozdílná doporučení péče pro nosiče variant konkordantních s fenotypem probanda a pro nosiče variant zachycených jako sekundární nález [12].
Při tvorbě konsenzu bylo rovněž diskutováno reportování monoalelických P/LP variant spojených s AR syndromy a s X-vázanými onemocněními u žen-přenašeček. Panovala shoda na rutinním reportování sekundárně zachycených P/LP variant v genech, v nichž frekvence heterozygotů v naší populaci přesahuje frekvenci 1/200 z důvodu prevence AR onemocnění. Frekvence heterozygotů v naší populaci byla stanovena na souboru 6 879 neselektovaných kontrol (1. LF UK a VFN v Praze) a 12 421 jedinců vyšetřených především z důvodů infertility, abortu či jiných než onkologických příčin (Gennet, Praha). V 95% konfidenčním intervalu zahrnujícím frekvenci heterozygotů v naší populaci 1/200 se v obou souborech pohybovaly následující geny spojené se závažnými AR syndromy: ATM, BLM, FANCA a NBN. U nosičů P/LP variant v těchto genech je doporučeno prekoncepční testování partnerů. Při záchytu P/LP varianty v genech FANCB a GPC3 spojených s X-vázanými onemocněními je u žen-přenašeček doporučeno genetické poradenství s cílem prevence X-vázaného onemocnění u mužských potomků.
Závěr
Problematika sekundárních nálezů a rozsahu reportování při genetickém vyšetření nádorové predispozice je velmi aktuálním tématem současné lékařské genetiky i preventivní onkologie. Technologický rozvoj přinesl vedle masivního nárůstu testování nádorové predispozice, jasného diagnostického zisku a rychlé odpovědní doby i výzvy spojené s interpretací a komunikací sekundárních nálezů. Absence jednotného postupu vedla v ČR k vytvoření odborného konsenzu v rámci konsorcia CZECANCA a pracovní skupiny Onkogenetika SLG JEP, který představuje důležitý krok směrem ke standardizaci péče o osoby s genetickou predispozicí k nádorovým onemocněním v ČR. Protože se jedná o rychle se rozvíjející problematiku, lze předpokládat, že s rostoucím množstvím znalostí se bude vyvíjet i rozsah reportovaných genů. Aktuálně platná doporučení vytvářená na základě diskuze široké odborné veřejnosti jsou dostupná na webových stránkách pracovní skupiny Společnosti lékařské genetiky [13].
Dedikace
Práce byla podpořena granty MZ ČR (RVO-VFN-64165 a NU22-03-00276) a MŠMT ČR (Program EXCELES, projekt číslo LX22NPO5102).
Sources
1. Spurdle AB, Greville-Heygate S, Antoniou AC et al. Towards controlled terminology for reporting germline cancer susceptibility variants: an ENIGMA report. J Med Genet 2019; 56 (6): 347–357. doi: 10.1136/jme dgenet-2018-105872.
2. Turnbull C, Achatz MI, Balmaña J et al. Breast cancer germline multigene panel testing in mainstream oncology based on clinical-public health utility: ESMO Precision Oncology Working Group recommendations. Ann Oncol 2025; 36 (8): 853865. doi: 10.1016/j.annonc.2025.04.012.
3. Michaan N, Leshno M, Safra T et al., Cost effectiveness of whole population BRCA genetic screening for cancer prevention in Israel. Cancer Prev Res (Phila) 2021; 14 (4): 455–462. doi: 10.1158/1940-6207.CAPR-20-0411.
4. Witt D, Sturm M, Stäbler A et al. Clinical genome sequencing in patients with hereditary breast and ovarian cancer: concept, implementation and benefits. Breast 2025; 82 : 104505. doi: 10.1016/j.breast.2025.
5. Lee K, Abul-Husn NS, Amendola LM et al. ACMG SF v3.3 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2025; 27 (8): 101454. doi: 10.1016/j.gim.2025.101454.
6. ACMG secondary finding genes and diseases. [online]. Available from: https: //search.clinicalgenome.org/kb/genes/acmgsf.
7. Gregg AR, Aarabi M, Klugman S et al. Screening for autosomal recessive and X-linked conditions during pregnancy and preconception: a practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2021; 23 (10): 1793–1806. doi: 10.1038/s41436-021-01203-z.
8. Schmitz MJ, Bashar A, Soman V et al. Leveraging diverse genomic data to guide equitable carrier screening: insights from gnomAD v.4.1.0. Am J Hum Genet 2025; 112 (1): 181–195. doi: 10.1016/j.ajhg.2024.11.004.
9. Czech Cancer Panel for Clinical Application. [online]. Available from: www.czecanca.cz
10. Gordon AS, Lee K, Abul-Husn NS et al. Consideration of disease penetrance in the selection of secondary findings gene-disease pairs: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2024; 26 (7): 101142. doi: 10.1016/j.gim.2024.101142.
11. Jackson L, Weedon MN, Green HD et al. Influence of family history on penetrance of hereditary cancers in a population setting. EClinicalMedicine 2023; 64 : 102159. doi: 10.1016/j.eclinm.2023.102159.
12. Skefos CM, Brock PL, Blouch E et al. SDHA secondary findings in germline testing: counseling and surveillance considerations. Endocr Oncol 2024; 4 (1): e230043. doi: 10.1530/EO-23-0043.
13. Onkogenetika. Pracovní skupina Společnosti lékařské genetiky (SLG). [online]. Dostupné z: www.onko - genetika.cz.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2026 Issue 1
Most read in this issue
- Rozsah reportování při vyšetření predispozice k nejčastějším solidním nádorům dospělého věku pomocí sekvenování nové generace
- Výsledky léčby Hodgkinova lymfomu v České republice
- Kožní nemoci způsobené monoklonálním imunoglobulinem a/nebo volnými lehkými řetězci (monoklonální gamapatie kožního významu)
- Logopedická intervence u pacientky s mnohočetným myelomem a dysfagií – případová studie z klinické praxe