
Změny v přístupu k analýze a hodnocení dědičných patogenních variant TP53
Authors:
B. Konečná 1; V. Krutílková 2; P. Kleiblová 1,3; L. Macůrek 4; Z. Kleibl 1,5
Authors‘ workplace:
Ústav lékařské biochemie a laboratorní diagnostiky 1. LF UK a VFN v Praze
1; Oddělení lékařské genetiky, Laboratoře AGEL a. s., Nový Jičín
2; Ústav biologie a lékařské genetiky 1. LF UK a VFN v Praze
3; Laboratoř biologie nádorových buněk, Ústav molekulární genetiky AV ČR v. v. i., Praha
4; Ústav patologické fyziologie 1. LF UK, Praha
5
Published in:
Klin Onkol 2025; 38(5): 358-367
Category:
Reviews
doi:
https://doi.org/10.48095/ccko2025358
Overview
Východiska: Liův-Fraumeniho syndrom (LFS) je vzácné autozomálně dominantní onemocnění charakterizované extrémním celoživotním rizikem vzniku mnohočetných a časně se objevujících zhoubných nádorů, za jehož vznik odpovídají dědičné patogenní mutace v genu TP53. Zatímco somatické mutace v TP53 patří mezi nejčastější genetické alterace u nádorových onemocnění, dědičné mutace jsou raritní. Poslední dekáda přinesla řadu změn v diagnostice LFS a nových poznatků o rizicích asociovaných nádorových onemocnění, které se promítají do preventivních opatření. Cíl: Předkládaný přehled ilustruje vývoj diagnostiky LFS a změny v indikačních kritériích pro testování germinálních variant v TP53, které umožnily definici širší nozologické jednotky – syndromu dědičné nádorové predispozice související s TP53 (hTP53rc – heritable TP53-related cancer syndrome). Posun v molekulárně biologické diagnostice směrem k sekvenování nové generace zjednodušil postup a dostupnost vyšetření nádorové predispozice. Analýza genu TP53 na germinální úrovni se stala součástí vyšetření téměř u všech osob indikovaných k testování dědičné nádorové predispozice, i když se nejedná o osoby s LFS fenotypem. Identifikace skutečných zárodečných patogenních variant TP53 představuje výzvu v jejich odlišení od mozaicizmu či somatických patogenních variant TP53 vznikajících v důsledku klonální hematopoézy neurčitého potenciálu. Pro jejich odlišení je nezbytná konfirmační analýza ze vzorku tkáně s vyjádřením alelické frakce variantní alely. Výzvou je i hodnocení patogenity vzácných germinálních variant TP53, která vyžaduje komplexní analýzu korelace genotypu s fenotypem. Pro potvrzení patogenity germinálních variant TP53 byla recentně publikována genově specifická kritéria American College of Medical Genetics and Genomics / Association for Molecular Pathology. Pouze nosiči jednoznačně prokázané zárodečné patogenní varianty by měli být zařazeni do intenzivního klinického preventivního programu. Závěr: Předložená práce upozorňuje na posun v poznání a diagnostice jedné z nejsilnějších predispozic ke vzniku nádorových onemocnění a měla by sloužit jako východisko pro další diskuzi týkající se organizace péče o vysoce rizikové nosiče patogenních variant TP53 nejen v dětském, ale i dospělém věku v ČR.
Klíčová slova:
hTP53rc – gen TP53 – nádorová predispozice – sekvenování nové generace (NGS) – klonální hematopoéza neurčitého potenciálu (CHIP) – mozaicizmus – Liův-Fraumeniho syndrom (LFS)
Úvod
Liův-Fraumeniho syndrom (LFS) je vzácné autozomálně dominantní onemocnění spojené s extrémním rizikem vzniku časných a nezřídka mnohočetných zhoubných nádorů. Celoživotní riziko vzniku nádorového onemocnění dosahuje u mužů s LFS 70 % a u žen téměř 100 % [1]. Příčinou LFS jsou germinální patogenní varianty v genu TP53 kódujícím tumor supresorový protein p53, který je transkripčním faktorem trvale exprimovaným ve všech buňkách. Do současné doby byly popsány stovky genů, jejichž expresi p53 ovlivňuje, vč. genů řídících řadu důležitých intracelulárních signálních drah kritických ve vztahu k tumorogenezi (obr. 1).


TP53 je nejčastěji postiženým genem ve zhoubných nádorech (ZN). S jeho v naprosté většině somatickými mutacemi se setkáváme u poloviny všech lidských nádorových onemocnění [2]. Oproti tomu jsou zárodečné mutace TP53 způsobující LFS velmi vzácné a v naší populaci nepřesahují četnost 1–3/100 000 [3]. Klasická forma LFS je klinicky definována přítomností typických nádorů, které zahrnují sarkomy měkkých tkání či osteosarkomy (bez Ewingova sarkomu), adrenokortikální karcinomy, tumory CNS a karcinomy prsu u mladých žen. Ke vzniku těchto onkologických onemocnění dochází typicky již v dětském věku či časné dospělosti (u ZN prsu). V souvislosti s LFS se častěji vyskytují také jinak vzácná nádorová onemocnění (hypodiploidní akutní lymfoblastická leukemie nebo tumory choroidálního plexu) (graf 1).
Zpočátku se analýza genu TP53 prováděla pouze u pacientů splňujících kritéria klasické formy LFS (viz dále). V současnosti je testování TP53 součástí většiny multigenových panelů pro diagnostiku hereditárních nádorových onemocnění, což umožnilo u nosičů zárodečných patogenních variant v TP53 identifikovat výrazně širší škálu fenotypů (onkologických onemocnění), než které odpovídají klasickým kritériím LFS (graf 1). Variabilita klinických projevů vedla k postupnému rozvolnění diagnostických kritérií a k zavedení širší nozologické jednotky označované jako syndrom dědičné nádorové predispozice související s TP53 (heritable TP53-related cancer syndrome – hTP53rc) [4]. Vlastní LFS je ve skutečnosti podmnožinou hTP53rc tvořenou těmi nosiči patogenních mutací TP53, kteří splňují klasická klinická kritéria LFS. V praxi (a literatuře) se často oba termíny používají promiskuitně.
V minulosti byly s LFS asociovány i zárodečné patogenní varianty v genu CHEK2, avšak postupné doplnění a rozšíření informací o nádorovém spektru u nosičů patogenních variant TP53 a CHEK2 jasně vylučuje, že by varianty v CHEK2 byly spojeny s LFS [5].
V následujícím textu se budeme věnovat významu a interpretaci nálezů patogenních variant v genu TP53 za různých okolností a postupům v interpretaci výsledků molekulárně genetické analýzy, které významně ovlivňují následnou onkologickou péči o vyšetřované osoby.
Diagnostická a indikační kritéria LFS

S ohledem na širokou škálu fenotypů pozorovaných v rodinách s LFS (a nověji u pacientů s hTP53rc) bylo v průběhu let vyvinuto několik různých verzí kritérií k identifikaci osob s LFS a následně nosičů patogenních variant TP53 (tab. 1). LFS byl jako syndrom popsán v roce 1969 [7], ale první, tzv. klasická kritéria LFS, byla představena o více než desetiletí později na základě vyhodnocení klinických charakteristik dostatečného počtu rodin s LFS [8]. Klasická kritéria byla využívána ke stanovení klinické diagnózy LFS bez ohledu na identifikaci patogenní zárodečné mutace TP53. Teprve následně bylo prokázáno, že LFS vzniká v důsledku nosičství dědičných patogenních variant genu TP53 [9]. V polovině 90. let představily Birchová a Eelesová širší kritéria pro identifikaci rodin, které odpovídají diagnóze LFS-like (LFL), jež zahrnuje pacienty nesplňující všechny „klasické“ znaky LFS [10,11]. V roce 2001 zavedli Chompretová et al. první indikační kritéria pro diagnostiku LFS s cílem zachytit jedince a rodiny se zárodečnými mutacemi TP53, kteří nemusí splňovat klasická kritéria, ale přesto mají vysoké riziko nádorových onemocnění [12]. Na základě robustní analýzy fenotypů nosičů germinálních mutací v TP53 Bougeardová et al. aktualizovali v roce 2008 kritéria Chompretové. Tato tzv. modifikovaná kritéria Chompretové umožnila lépe identifikovat rodiny s mírnějšími fenotypy [12,13].
Zatím poslední významná revize kritérií Chompretové provedená v roce 2015 zohledňuje pokroky v molekulárně genetické diagnostice s důrazem na genetické testování pro identifikaci jedinců s dědičnou mutací TP53 bez typické rodinné anamnézy LFS (tedy pacienty s hTP53rc). Tato tzv. revidovaná kritéria Chompretové rozšiřují spektrum nádorů spojených s LFS (vč. dětských nádorů choroidálního plexu) a zahrnují klinická doporučení pro nosiče mutací TP53 s cílem optimalizovat strategie prevence a časného záchytu nádorových onemocnění [6].
Klasická kritéria se vyznačují vysokou pozitivní prediktivní hodnotou a vysokou specifičností (50–70 % v některých studiích), avšak poměrně nízkou senzitivitou (kolem 40 %) vyplývající ze skutečnosti, že u jedinců s patogenními variantami TP53 se mohou vyvinout nádorová onemocnění mimo nádory typické pro LFS (obr. 2). Aplikací pouze klasických LFS kritérií je tedy možné minout řadu nosičů patogenních variant. Pozdější kritéria vykazují výrazně nižší specifičnost (kritéria Chompretové 20–35 %), avšak spojením klasických a revidovaných kritérií LFS je možné dosáhnout až 95% záchytnosti patogenních variant v TP53 [14].



genní varianta, VAF – variantní alelická frakce
Indikační kritéria pro testování osob se zvýšeným rizikem přítomnosti dědičných variant v TP53 se mírně odlišují v různých zemích, což odráží vývoj poznání, ale i regionální preference, dostupné zdroje a konsenzus odborníků v rámci národní lékařské komunity. Doporučení pro testování germinálních mutací TP53 byla v ČR poprvé publikována v roce 2012 [16]. Rozšířená verze indikačních kritérií vyšla v rámci knihy Hereditární nádorová onemocnění v klinické praxi v roce 2022 [17] a aktualizována byla výborem Společnosti lékařské genetiky v roce 2024 (tab. 2). V souladu se současnými mezinárodními doporučeními lze o nosičství dědičné patogenní varianty v TP53 uvažovat i u pacientů mladších 46 let, u kterých se vyvine sekundární nádorové onemocnění v místě ozařování primárního ložiska nádoru typického pro diagnózu LFS [4]. Tito pacienti jsou však již dnes v ČR indikováni ke germinálnímu genetickému testování.
Patogenní varianty v genu TP53
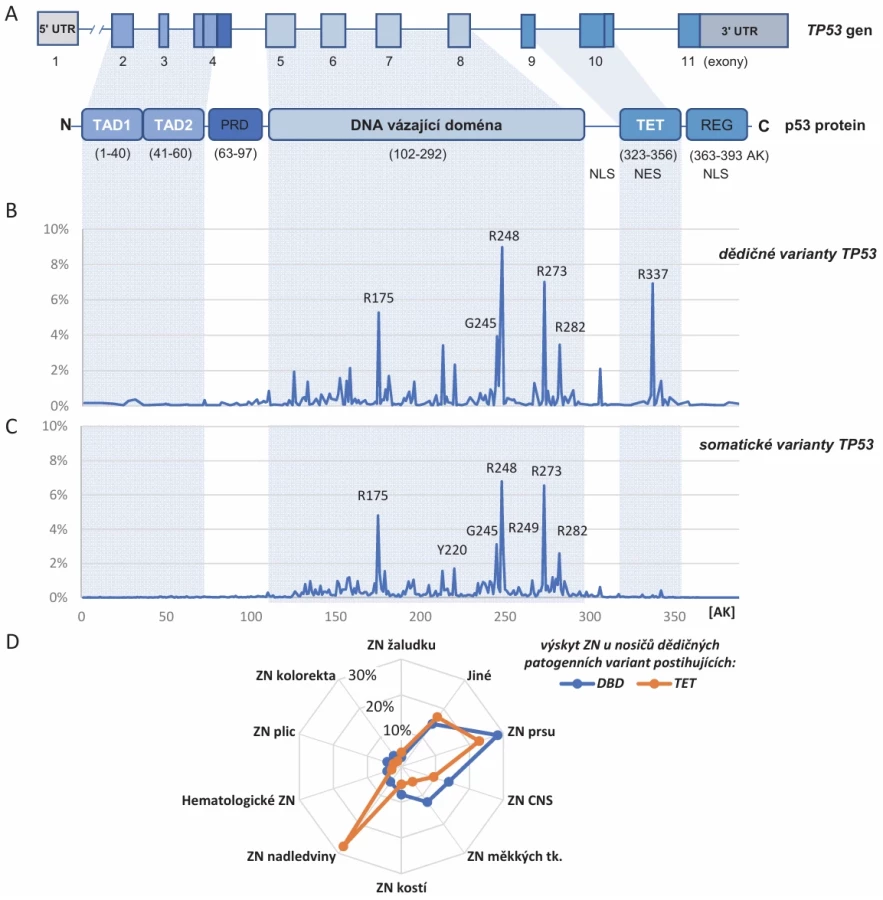
Gen TP53 je lokalizován na krátkém raménku chromozomu 17 (17p13.1), kde zahrnuje oblast o velikosti ~20 kb (17p13.1). Jeho kanonický transkript (NM_000546.5) obsahuje 11 exonů, z nichž 10 kóduje proteinový produkt p53 o velikosti 393 aminokyselin (obr. 2). Genetické testování zárodečných variant v genu TP53 lze rozdělit na dvě základní indikace. První je testování přítomnosti dědičné mutace v TP53 u osoby s typickým nádorovým fenotypem LFS. Druhým, dnes značně častějším případem je zjištění přítomnosti patogenní varianty TP53 v průběhu genetického testování zárodečné DNA pomocí panelového, celoexomového či celogenomového sekvenování nové generace (next generation sequencing – NGS) u onkologického pacienta či u indikovaného jedince bez onkologického onemocnění.
Klasifikace identifikovaných dědičných variant TP53 je založena na pětitřídním systému dle American College of Medical Genetics and Genomics (ACMG) / Association for Molecular Pathology (AMP), podobně jako klasifikace germinálních variant v ostatních nádorových predispozičních genech [18,19]. S ohledem na závažnost diagnózy LFS je však hodnocení zárodečných variant specificky uzpůsobeno modifikací obecných doporučení na míru genu TP53 [20]. Hlavní změny se týkají hodnocení vzácných missense variant, u kterých je pro zařazení mezi patogenní (class 5) nebo pravděpodobně patogenní (class 4) varianty vyžadováno funkční vyšetření pomocí hodnověrných in vitro testů. Dosud byly publikovány tři rozsáhlé systematické funkční studie missense variant TP53 hodnotící transaktivační aktivitu [21], ztrátu funkce [22,23] a dominantně negativní účinek variantních izoforem p53 [22].
Tradičním zdrojem informací o variantách v TP53 je TP53 Database [24], která agreguje publikovaná výzkumná i klinická data od 90. let 20. století (nejprve pod hlavičkou International Agency for Research on Cancer (IARC) a WHO, nyní pod správou National Cancer Institute (NCI)) [25]. Referenčním zdrojem pro klasifikaci germinálních variant způsobujících lidská onemocnění vč. variant v TP53 ve vztahu k LFS/hTP53rc je databáze ClinVar [26]. K 29. 4. 2025 bylo v databázi ClinVar zařazeno 3 615 různých germinálních variant v genu TP53, z nichž více než třetina je klasifikována jako patogenní nebo pravděpodobně patogenní varianty způsobující ztrátu fyziologické funkce proteinu p53 a jsou považovány za příčinné pro vznik LFS/hTP53rc. Další třetinu tvoří skupina benigních (class 1) a pravděpodobně benigních (class 2) alterací (neutrálních variant bez vztahu k LFS/hTP53rc) a poslední část (kolem 30 % všech) tvoří varianty nejasného významu (variants of uncertain significance – VUS), class 3, u kterých souvislost s onemocněním v současnosti nelze ani prokázat, ani vyloučit. Většinu patogenních (class 4–5) alterací tvoří mutace vedoucí ke vzniku zkráceného proteinu p53 (delece/inzerce, nonsense nebo sestřihové varianty). Pouze pětina (220/1098) ze všech typů reportovaných pravděpodobně patogenních či patogenních variant TP53 představují missense varianty, avšak tyto se vyskytují zdaleka nejčastěji a jsou přítomné u přibližně 70 % osob s LFS (obr. 2B). Missense varianty tvoří rovněž většinu patogenních alterací v případě somatických mutací (obr. 2C). Spektrum germinálních variant v genu TP53 je tedy velmi pestré a do značné míry se překrývá se somatickými variantami přítomnými v nádorových buňkách.
Alterace klíčových strukturních aminokyselinových zbytků proteinu p53 v důsledku somatických či dědičných variant jsou patrné jako tzv. hot spot mutace. Ty se kumulují především v oblasti DNA-vázající domény (DBD; obr. 2), kde postihují aminokyseliny zodpovědné za interakci transkripčního faktoru p53 s DNA nebo strukturní integritu DBD. Třebaže jsou hot spot mutace nápadně časté, tvoří dohromady přibližně pouze třetinu ze všech patogenních variant. Přítomnost dědičných mutací postihujících DBD je při srovnání proti patogenním variantám lokalizovaným mimo tuto doménu spojena s vyšší penetrancí, nižším věkem vzniku nádorového onemocnění a vyšším stupněm malignity [6]. Nejčastější missense zárodečná mutace mimo DBD je populačně specifická „brazilská“ hypomorfní (pouze částečně nefunkční) varianta p.R337H [27]. Nosiči této varianty mají poněkud odlišné spektrum asociovaných zhoubných nádorů s vysokým rizikem vzniku adrenokortikálních tumorů (obr. 2D), ale výrazně méně často dalších typických nádorů pro LFS. Ženy nosičky p.R337H mají přibližně 90% celoživotní riziko karcinomu prsu, ale významně nižší výskyt onemocnění v mladém věku. Uvádí se, že u méně než 20 % pacientů s p.R337H se vyvine onkologické onemocnění ve věku do 30 let, zatímco u nosičů ostatních patogenních TP53 variant je to kolem 50 % [28–30].
Některé z izoforem missense variant vykazují dominantně negativní účinek, kdy vznikající mutované formy tetramerních p53 proteinů mohou transkripčně aktivovat i zcela jiné geny než wild-type p53 [29,31,32]. Důkazy o dominantně negativním působení dědičných patogenních variant (např. R175H, R246S a R270H) pocházejí však především z in vitro nebo in vivo experimentů na zvířecích modelech a jejich význam s ohledem na osoby s LFS není klinicky zcela zřejmý [33].
V naší populaci bylo popsáno celé spektrum patogenních variant TP53 vč. delecí celého genu [34], proto je pro vyloučení patogenních variant v TP53 žádoucí věnovat pozornost i strukturním přestavbám (CNV). Nejčastější dědičné mutace TP53 kopírují rozložení uvedené v obr. 2B, hypomorfní varianta p.R337H se však v ČR nativně nevyskytuje.
Úskalí interpretace nálezů zárodečných patogenních variant v TP53
Identifikace a následná interpretace vzácných genetických alterací TP53 vyžaduje komplexní přístup zahrnující hodnocení genetické (výskyt variant v různých populacích a populačních databázích, segregace varianty s onemocněním v postižených rodinách), molekulárně biologické (funkční in vitro analýzy), patologické (identifikace ztráty funkční alely v nádorech u nosičů) a klinické (asociace se vznikem typických/atypických onkologických diagnóz a jejich subtypů) [20]. Komplikaci v diagnostice germinálních patogenních variant v TP53 představuje především klonální hematopoéza neurčitého potenciálu (clonal hematopoiesis of indeterminate potential – CHIP), tj. klonální expanze leukocytů z prekurzorových buněk kostní dřeně, které v průběhu života vyvinuly somatickou mutaci TP53. S ohledem na obrovská rizika vzniku zhoubných nádorů u nosičů zárodečných mutací v TP53 a s tím spojený intenzivní klinický management je nezbytné, abychom v rámci diagnostiky správně určili, zda se v případě přítomnosti patogenní varianty při vyšetření z krve jedná o skutečnou zárodečnou patogenní variantu v heterozygotním stavu, nebo mozaiku, případně zda se u vyšetřovaného varianta v TP53 vyskytuje v důsledku CHIP. Gen TP53 patří mezi pětici nejčastěji postižených genů asociovaných s výskytem CHIP [35]. Odhaduje se, že až 20 % komerčních laboratorních zpráv v USA referujících přítomnost dědičné patogenní varianty v TP53 ve skutečnosti představuje nejspíše CHIP, a nikoliv skutečný záchyt zárodečné mutace [36].
Heterozygotní germinální patogenní varianta by měla být přítomna ve všech somatických buňkách (s výjimkou patologických nádorových buněk) s konzistentní frakcí variantní alely blízkou 50 % (obr. 3). Je tedy vždy nezbytné, aby genetická zpráva o zachycení dědičné mutace TP53 byla doplněna údajem o alelické frakci variantní alely v primárně vyšetřovaném vzorku (obvykle krvi). Nicméně ani 50 % zastoupení variantní alely ve vzorku krve nevylučuje přítomnost CHIP. Pro potvrzení skutečného zárodečného původu je zároveň nutné potvrzení přítomnosti patogenní varianty v alternativní tkáni buněk nehematopoetického původu, např. z bukální sliznice nebo optimálně z vlasových folikulů (obr. 4), opět se stanovením její alelické frakce. Vyšetření z bukálního stěru je však méně vhodné z důvodu možné kontaminace leukocyty. Alternativou potvrzení dědičného původu je prokázání přítomnosti stejné příčinné mutace u biologického příbuzného. Zde je přitom nezbytné vzít v úvahu, že patogenní varianta TP53 může vzniknout u některých (~10 %) pacientů s LFS de novo v rámci gametogeneze [37]. Nízká frakce variantní alely TP53 (< 30 %) v primárním vzorku i ve vzorku další analyzované tkáně značí přítomnost vzácného postzygotického mozaicizmu, kdy mutace v genu TP53 vzniká v některé z buněk blastomery v rámci časné embryogeneze (obr. 3B). Při výskytu CHIP (obr. 3C) je obvyklá přítomnost alterace TP53 při analýze zárodečné DNA ze vzorku krve, která není detekovatelná při analýze další tkáně. Pravděpodobnost CHIP výrazně stoupá u osob starších 50 let nebo u osob po systémové chemoterapii [38]. U jedinců starších 65 let lze CHIP detekovat v 10 % všech případů analýzy germinální DNA z krve [39].
Přítomnost dědičné patogenní varianty indikuje rovněž konkordantní klinický nález, ve kterém by měly dominovat klasické tumory asociované s LFS. Komplikací je identifikace zárodečné patogenní varianty u osoby bez známé rodinné anamnézy (např. cizinci, osvojené osoby apod.), kde se v rámci hTP53rc může vyskytnout v podstatě libovolná onkologická diagnóza, i když pravděpodobnost nosičství příčinné patogenní varianty významně klesá s věkem (a po 60. roce věku je nepravděpodobná). Všechny osoby s prokázanou dědičnou patogenní variantou v genu TP53 jsou indikovány k intenzivnímu klinickému sledování a kaskádové analýze mutace u přímých příbuzných bez ohledu na pohlaví a věk (vč. dětí).
V případě potvrzeného mozaicizmu TP53 je doporučeno klinické sledování jako u osob s diagnózou LFS, včetně prediktivního testování potomků nosiče.
Jak bylo uvedeno výše, přítomnost CHIP se zvyšuje v souvislosti s rostoucím věkem. Na eventualitu CHIP je dobré myslet vždy, když je detekována jasná patogenní varianta v TP53 (vč. hot spot mutací) bez uvedení variantní alelické frakce, především u starší dospělé osoby bez zjevného onkologického onemocnění a/nebo bez nápadné rodinné onkologické anamnézy. U osob s prokázaným CHIP zatím nejsou klinická doporučení pro sledování jasně stanovena, neboť rizika rozvoje hematologických malignit (nejčastěji myeloidního, ale i lymfoidního původu) či asociovaných nehematologických onemocnění (ICHS, diabetu mellitu 2. typu) nebyla doposud vyčíslena. Studie z Mayo Clinic doporučuje retestování CHIP u osob s přetrvávající (≥ 4 měsíce) nevysvětlitelnou cytopenií, u pacientů s malignitami před adjuvantní cytotoxickou chemoterapií a/nebo radiační léčbou, před autologní transplantací krvetvorných kmenových buněk a CAR-T terapií [40].
Závěr
Interpretace genetického nálezu je v případě genu TP53 při vyšetření zárodečné DNA specifickým problémem vyžadujícím pečlivý přístup pro vyhodnocení klinického významu nalezené varianty i dodržení doporučeného postupu, vč. konfirmačního vyšetření z nezávislého vzorku nehematopoetické tkáně (obr. 4) pro identifikaci germinální podstaty varianty a jejího odlišení od mozaicizmu nebo CHIP. Vysoká penetrance germinálních patogenních variant tumor supresorového genu TP53 zdůrazňuje jeho význam v onkogenezi a odůvodňuje smysl intenzivních preventivních opatření, která jsou nezbytnou podmínkou zlepšení prognózy u nosičů – pacientů s LFS nebo hTP53rc. Na druhou stranu, vysoká intenzita prevence (zaměřené na časnou detekci nádorů prakticky v libovolné lokalizaci) vyžaduje, aby do sledovacích programů byli zařazeni pouze nosiči skutečných prokázaných zárodečných patogenních/pravděpodobně patogenních variant (class 4 a 5) v TP53, a nikoliv nosiči dědičných variant nejasného významu (class 3), nepatogenních variant (class 1 a 2) nebo nosiči patogenních nezárodečných variant v důsledku CHIP.
Předkládaný text si klade za cíl vyvolat širší diskuzi na téma správné identifikace osob s LFS nebo hTP53rc, a tím i následné péče o ně v ČR. Zatímco péče o dětské pacienty se soustřeďuje především ve FN Motol, péče o dospělé nosiče mutací, vč. těch, u kterých byla identifikována patogenní varianta v TP53 v průběhu panelového vyšetření jako (v podstatě) vedlejší nález, v současnosti centralizována v ČR není. Na širší odborné diskuzi ponecháváme možnost zřízení centralizované péče o dospělé pacienty s LFS/hTP53rc a rovněž její konsenzuální aktualizaci napříč zainteresovanými zdravotnickými odbornostmi.
Podporující agentury a sponzoři: Tato práce byla podpořena Agenturou pro zdravotnický výzkum Ministerstva zdravotnictví ČR (NU22-03-00276 a DR-VFN-64165); Grantovou agenturou Karlovy Univerzity (SVV 260631, Cooperatio a UNCE 24/MED/022).
Sources
1. Chompret A. The Li-Fraumeni syndrome. Biochimie 2002; 84 (1): 75–82. doi: 10.1016/s0300-9084 (01) 01361-x.
2. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2010; 2 (1): a001008. doi: 10.1101/cshperspect.a001008.
3. Tinka P, Pavelka Z, Vejmělková K et al. Extrémně vzácn spinální nádor na podkladě Li-Fraumeni syndromu. Oncology 2023; 17 (5): 304–307. doi: 10.36290/xon.2023.056.
4. Frebourg T, Lagercrantz SB, Oliveira C et al. Guidelines for the Li-Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet 2020; 28 (10): 1379–1386. doi: 10.1038/s41431-020-0638-4.
5. Fortuno C, Richardson M, Pesaran T et al. CHEK2 is not a Li-Fraumeni syndrome gene: time to update public resources. J Med Genet 2023; 60 (12): 1215–1217. doi: 10.1136/jmg-2023-109464.
6. Bougeard G, Renaux-Petel M, Flaman JM et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol 2015; 33 (21): 2345–2352. doi: 10.1200/JCO.2014.59.5728.
7. Li FP, Fraumeni JF. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 1969; 71 (4): 747–752. doi: 10.7326/0003-4819-71-4-747.
8. Li FP, Fraumeni JF, Mulvihill JJ et al. A cancer family syndrome in twenty-four kindreds. Cancer Res 1988; 48 (18): 5358–5362.
9. Malkin D, Li FP, Strong LC et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250 (4985): 1233–1238. doi: 10.1126/science.1978757.
10. Birch JM, Hartley AL, Tricker KJ et al. Prevalence and diversity of constitutional mutations in the p53 gene among 21 Li-Fraumeni families. Cancer Res 1994; 54 (5): 1298–1304.
11. Eeles RA. Germline mutations in the TP53 gene. Cancer Surv 1995; 25 : 101–124.
12. Chompret A, Abel A, Stoppa-Lyonnet D et al. Sensitivity and predictive value of criteria for p53 germline mutation screening. J Med Genet 2001; 38 (1): 43–47. doi: 10.1136/jmg.38.1.43.
13. Bougeard G, Sesboüé R, Baert-Desurmont S et al. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet 2008; 45 (8): 535–538. doi: 10.1136/jmg.2008.057570.
14. Daly MB, Pal T, Maxwell KN et al. NCCN Guidelines Insights: genetic/familial high-risk assessment: breast, ovarian, and pancreatic, version 2.2024. J Natl Compr Canc Netw 2023; 21 (10): 1000–1010. doi: 10.6004/jnccn.2023.0051.
15. Tinat J, Bougeard G, Baert-Desurmont S et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol 2009; 27 (26): e108–e109. doi: 10.1200/JCO.2009.22.7967.
16. Foretová L, Stěrba J, Opletal P et al. Li-Fraumeni syndrome – a proposal of complex prevention care for carriers of TP53 mutation with total-body MRI. Klin Onkol 2012; 25 (Suppl): S49–S54.
17. Foretová L, Macháčková E, Gaillyová R et al. Hereditární nádorová onemocnění v klinické praxi. Praha: Grada 2022.
18. Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17 (5): 405–424. doi: 10.1038/gim.2015.30.
19. Janatová M, Chvojka Š, Macháčková E et al. Classification of germline variants identified in cancer predisposition genetic testing – consensus of the CZECANCA consortium. Klin Onkol 2023; 36 (6): 431–439. doi: 10.48095/ccko2023431.
20. Fortuno C, Lee K, Olivier M et al. Specifications of the ACMG/AMP variant interpretation guidelines for germline TP53 variants. Human Mutation 2021; 42 (3): 223–236. doi: 10.1002/humu.24152.
21. Kato S, Han SY, Liu W et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A 2003; 100 (14): 8424–8429. doi: 10.1073/pnas.14316 92100.
22. Giacomelli AO, Yang X, Lintner RE et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 2018; 50 (10): 1381–1387. doi: 10.1038/s41588-018-0204-y.
23. Kotler E, Shani O, Goldfeld G et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell 2018; 71 (1): 178–190.e8. doi: 10.1016/j.molcel.2018.06.012.
24. The TP 53 database. [online]. Available from: https: //TP53.cancer.gov/.
25. de Andrade KC, Lee EE, Tookmanian EM et al. The TP53 database: transition from the International Agency for Research on Cancer to the US National Cancer Institute. Cell Death Differ 2022; 29 (5): 1071–1073. doi: 10.1038/s41418-022-00976-3.
26. ClinVar Database. [online]. Available from: https: //www.ncbi.nlm.nih.gov/clinvar/.
27. Šmardová K, Koptíková J. Brazilian story of the R337H p53 mutation. Klin Onkol 2014; 27 (4): 247–254. doi: 10.14735/amko2014247.
28. Andrade KC, Santiago KM, Fortes FP et al. Early-onset breast cancer patients in the South and Southeast of Brazil should be tested for the TP53 p.R337H mutation. Genet Mol Biol 2016; 39 (2): 199–202. doi: 10.1590/1678-4685-GMB-2014-0343.
29. Amadou A, Achatz MIW, Hainaut P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li-Fraumeni syndrome. Curr Opin Oncol 2018; 30 (1): 23–29. doi: 10.1097/CCO.0000000000000423.
30. Robles AI, Harris CC. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol 2010; 2 (3): a001016. doi: 10.1101/cshperspect.a001016.
31. Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene 2007; 26 (15): 2202–2211. doi: 10.1038/sj.onc.1210294.
32. Monti P, Perfumo C, Bisio A et al. Dominant-negative features of mutant TP53 in germline carriers have limited impact on cancer outcomes. Mol Cancer Res 2011; 9 (3): 271–279. doi: 10.1158/1541-7786.MCR-10-0496.
33. Gencel-Augusto J, Lozano G. p53 tetramerization: at the center of the dominant-negative effect of mutant p53. Genes Dev 2020; 34 (17–18): 1128–1146. doi: 10.1101/gad.340976.120.
34. Foretová L, Sedláček Z, Křepelová A et al. Syndrom Li-Fraumeni – diagnostické a preventivní možnosti. Kazuistika pacientky s delecí celého genu TP53. 2010 [online]. Dostupné z: https: //www.linkos.cz/lekar-a-multidisciplinarni-tym/kongresy/po-kongresu/databaze-tuzemskych-onkologickych-konferencnich-abstrakt/syndrom-li-fraumeni-diagnosticke-a-preventivni-moznosti-kazuistika-pacientky-s-d/.
35. Steensma DP, Bejar R, Jaiswal S et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015; 126 (1): 9–16. doi: 10.1182/blood-2015-03-631747.
36. Weitzel JN, Chao EC, Nehoray B et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med 2018; 20 (8): 809–816. doi: 10.1038/gim. 2017.196.
37. Renaux-Petel M, Charbonnier F, Théry JC et al. Contribution of de novo and mosaic TP53 mutations to Li-Fraumeni syndrome. J Med Genet 2018; 55 (3): 173–180. doi: 10.1136/jmedgenet-2017-104976.
38. Prochazkova K, Pavlikova K, Minarik M et al. Somatic TP53 mutation mosaicism in a patient with Li-Fraumeni syndrome. Am J Med Genet A 2009; 149A (2): 206–211. doi: 10.1002/ajmg.a.32574.
39. Genovese G, Kähler AK, Handsaker RE et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371 (26): 2477–2487. doi: 10.1056/NEJMoa1409405.
40. Mangaonkar AA, Patnaik MM. Clonal hematopoiesis of indeterminate potential and clonal cytopenias of undetermined significance: 2023 update on clinical associations and management recommendations. Am J Hematol 2023; 98 (6): 951–964. doi: 10.1002/ajh.26915.
41. Pecháčková S, Burdová K, Macůrek L. WIP1 phospahates as pharmacological target in cancer therapy. [online]. Available from: https: //link.springer.com/article/10.1007/s00109-017-1536-2.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2025 Issue 5
Most read in this issue
- Monoklonální gamapatie klinického významu – nový skupinový název pro choroby způsobené monoklonálním imunoglobulinem a/nebo volnými lehkými řetězci. Změna přístupu k nemaligním gamapatiím
- Změny v přístupu k analýze a hodnocení dědičných patogenních variant TP53
- Výživa pacientov podstupujúcich transplantáciu hematopoetických kmeňových buniek
- Precizní medicína v hematoonkologii – léčba refrakterního mnohočetného myelomu s masivním extramedulárním postižením BRAF/MEK inhibitory