
Bartterov syndróm u detí: súbor ôsmich prípadov z Českej republiky a Slovenska
Authors:
Csomó Daniel 1; Papež Jan 2; Doležel Zdeněk 2; Dluholucký Martin 3; Sládková Eva 4; Podracká Udmila 1
Authors‘ workplace:
Detská klinika, Lekárska fakulta, Univerzita Komenského a Národný ústav detských chorôb, Bratislava
1; Pediatrická klinika, Lékařská fakulta, Masarykova univerzita a FN Brno
2; II. Detská klinika, Slovenská zdravotnícka univerzita a Detská fakultná nemocnica s poliklinikou, Banská Bystrica
3; Dětská klinika, FN Plzeň
4
Published in:
Čes-slov Pediat 2024; 79 (2): 102-107.
Category:
Original Papers
doi:
https://doi.org/10.55095/CSPediatrie2024/015
Overview
Bartterov syndróm zahŕňa skupinu vzácnych geneticky podmienených tubulopatií sprevádzaných zvýšenými močovými stratami solí. Patogenetickým podkladom je porucha transportných systémov zodpovedných za reabsorpciu solí predovšetkým v hrubom segmente vzostupného ramienka Henleho kľučky. Medzi základné charakteristiky Bartterovho syndrómu patrí hypokaliemická, hypochloremická metabolická alkalóza a sekundárny (hyperrenínový) hyperaldosteronizmus pri normálnom alebo nízkom systémovom krvnom tlaku. Klinické prejavy sa líšia v závislosti od postihnutého génu, pričom rozoznávame 5 rôznych génovo-špecifických fenotypov ochorenia. V článku uvádzame súbor 8 pacientov z Českej republiky a Slovenska s geneticky potvrdeným Bartterovým syndrómom a ich klinický a laboratórny fenotyp.
Klíčová slova:
tubulopatie – Bartterov syndróm – hypokaliémia – metabolická alkalóza – hyperaldosteronizmus
Úvod
Bartterov syndróm patrí k vzácnym dedičným tubulopatiám s masívnymi renálnymi stratami solí. V závislosti od typu postihnutého génu rozlišujeme 5 základných typov Bartterovho syndrómu (BS1-5). Dedičnosť je autozómovo-recesívna pri BS1-4 a X-viazaná recesívna pri BS5.(1) Celková prevalencia všetkých typov Bartterovho syndrómu v Severnej Amerike a západoeurópskych krajinách sa odhaduje na 1 : 40 000 až 1 : 50 000.(2)
Génový defekt vedie k masívnym stratám solí močom a aktivácii renín-angiotenzín-aldosterónového systému s následným rozvojom hypokaliemickej, hypochloremickej metabolickej alkalózy. Patofyziologicky ide o poruchu tubuloglomerulovej spätnej väzby, kedy v bunkách macula densa dochádza k nadmernej aktivácii cyklooxygenáz (predovšetkým COX-2) a tvorbe prostaglandínov, najmä prostaglandínu E2.(1,3) Tubulárna dysfunkcia pri Bartterovom syndróme vedie aj k poruche spätnej resorpcie vápnika s hyperkalciúriou a progresívnou medulárnou nefrokalcinózou(4) a tiež k zníženiu či úplnému vymiznutiu osmotického gradientu v obličkovej dreni s následnou poruchou koncentračnej schopnosti obličiek.(5)
Klinické prejavy ochorenia, vek ich nástupu, ale aj stupeň biochemických abnormalít sa líšia podľa typu postihnutého génu (tab. 1, obr. 1). BS3 sa označuje aj ako klasický typ so zvyčajnou klinickou manifestáciou v prvých rokoch života, kým ostatné typy predstavujú antenatálne formy ochorenia. Klinický obraz môže zahŕňať polyhydramnion, predčasný pôrod, polyúriu a polydipsiu, prejavy hypovolémie, rekurentné vracanie, opakované horúčky, neprospievanie či poruchu rastu.(1,6) Napriek tomu, že pri BS5 sa spomedzi všetkých typov vyskytuje najzávažnejší polyhydramnion, polyúria sa zvyčajne spontánne upraví v priebehu prvých týždňov, najneskôr počas prvých dvoch rokov života.(7) Deti s BS4a a 4b majú aj senzorineurálnu poruchu sluchu, ktorá sa zistí skríningovým vyšetrením otoakustických emisií.(8)
Diagnostika Bartterovho syndrómu sa opiera o klinické prejavy a charakteristické laboratórne nálezy, ako je hypokaliemická, hypochloremická metabolická alkalóza, zvýšené koncentrácie renínu a aldosterónu pri normálnom či nízkom tlaku krvi, izostenúria, zvýšené frakčné exkrécie chloridov, prípadne hyperkalciúria a sonografický nález medulárnej nefrokalcinózy pri niektorých typoch ochorenia.(6)
Terapia pozostáva zo suplementácie draslíka a NaCl. Cieľom je udržať hodnoty kaliémie na ≥ 3 mmol/l, aby nedošlo k obávaným prejavom hypokaliémie.(6) U symptomatických pacientov, ktorí nedostatočne odpovedajú na substitúciu iónov, sa pristupuje k liečbe nesteroidnými antiflogistikami, ktoré potláčajú nadmernú tvorbu prostaglandínu E2.(1)
Napriek významným pokrokom v patogenéze sú dostupné údaje o genotypovo-fenotypových charakteristikách a dlhodobej prognóze ochorenia limitované. V nasledujúcom článku prezentujeme komplexné údaje česko-slovenskej kohorty 8 pacientov s Bartterovým syndrómom.
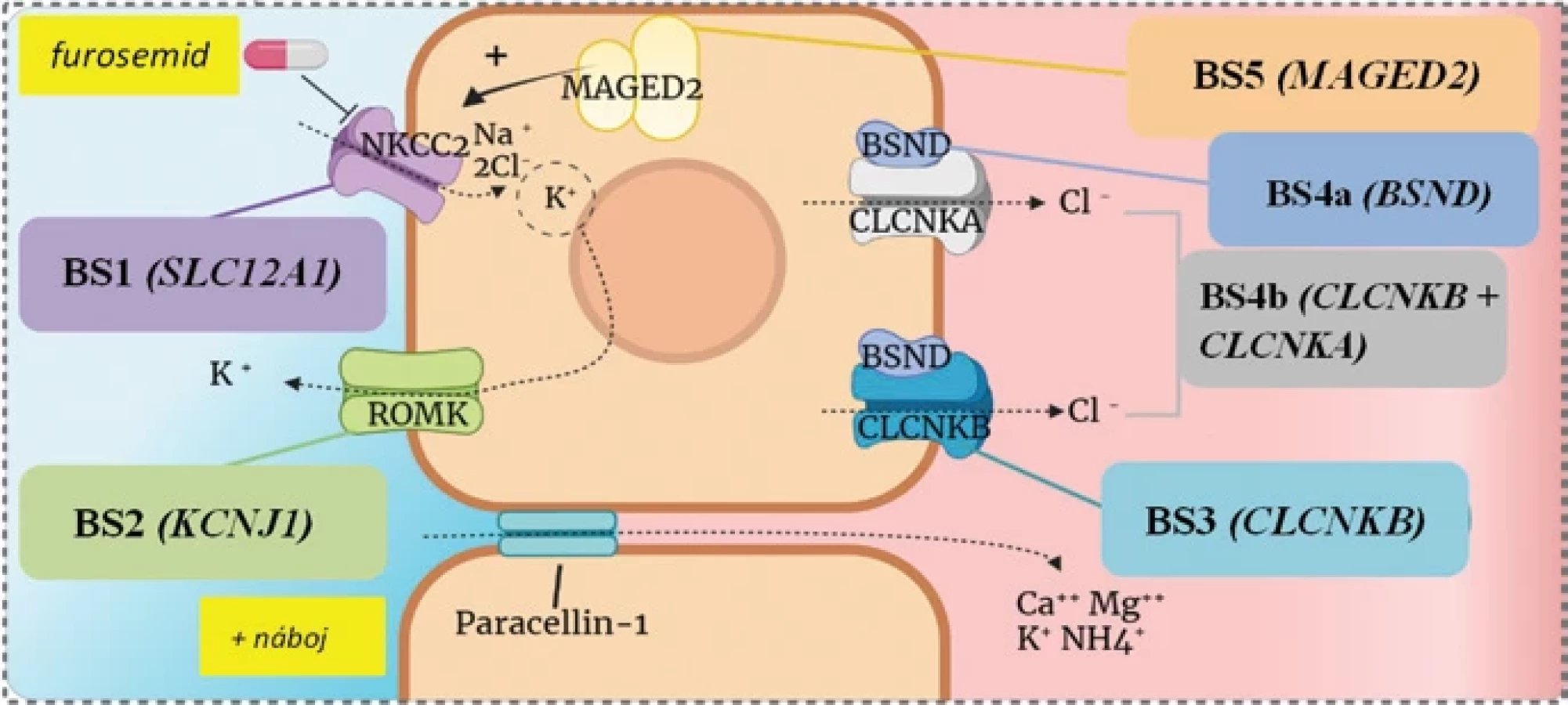
Tab. 1: Niektoré zo základných charakteristík jednotlivých typov Bartterovho syndrómu, upravené podľa(6)
|
|
BS1 |
BS2 |
BS3 |
BS4a |
BS4b |
BS5 |
|
Gén/dedičnosť |
SLC12A1/AR |
KCNJ1/AR |
CLCNKB/AR |
BSND/AR |
CLCNKA + CLCNKB/AR |
MAGED2/XR |
|
Prvá manifestácia |
prenatálne |
prenatálne |
0 – 5 rokov |
prenatálne |
prenatálne |
prenatálne |
|
Polyhydramnion |
závažný |
závažný |
neprítomný/mierny |
závažný |
závažný |
veľmi závažný |
|
Prematurita |
častá |
častá |
vzácna |
častá |
častá |
vždy |
|
Neonatálna |
častá |
častá |
vzácna |
častá |
častá |
častá |
|
Hypokaliémia |
vždy |
vždy |
vždy |
vždy |
vždy |
vždy |
|
Exkrécia vápnika |
vysoká |
vysoká |
variabilná, zvyčajne normálna |
variabilná |
variabilná |
variabilná |
|
Nefrokalcinóza |
veľmi častá |
veľmi častá |
vzácna/mierna |
vzácna/mierna |
vzácna/mierna |
vzácna |
|
Exkrécia horčíka |
normálna |
normálna |
normálna až vysoká |
normálna až vysoká |
normálna až vysoká |
neznáma |
|
Neprospievanie, porucha rastu |
bežné |
bežné |
bežné |
bežné |
bežné |
nie |
|
Iné prejavy |
|
prechodná novorodenecká hyperkaliémia |
|
senzorineurálna porucha sluchu |
senzorineurálna porucha sluchu |
large for gestational age; tranzietné ochorenie |
BS1 – BS5 – Bartterov syndróm 1. až 5. typ; AR – autozómovo-recesívna dedičnosť; XR – X-viazaná recesívna dedičnosť
Tab. 2: Vybrané charakteristiky jednotlivých pacientov s Bartterovým syndrómom. Klinické charakteristiky, prítomnosť nefrokalcinózy a hodnoty laboratórnych a antropometrických parametrov sú uvádzané v čase manifestácie ochorenia. Hodnota eGFR zodpovedá kalkulácii podľa aktuálnych parametrov pri poslednej ambulantnej kontrole.
|
|
Pacient 1 |
Pacient 2 |
Pacient 3 |
Pacient 4 |
Pacient 5 |
Pacient 6 |
Pacient 7 |
Pacient 8 |
|
Pohlavie |
ženské |
mužské |
mužské |
ženské |
ženské |
ženské |
mužské |
mužské |
|
Typ Bartterovho syndrómu |
III. |
III. |
III. |
I. |
II. |
III. |
III. |
I. |
|
Gén |
CLCNKB |
CLCNKB |
CLCNKB |
SLC12A1 |
KCNJ1 |
CLCNKB |
CLCNKB |
SLC12A1 |
|
Patogénny variant |
homozygot: c.1270G>A, p.Gly424Arg |
homozygotná delécia celého génu |
zložený heterozygot: Ex1-Ex19del; c.1271G>A, p.Gly424Glu |
zložený heterozygot: c.1522G>A, p.Ala508Thr; c.1675A>T, p.Ile559Phe |
zložený heterozygot: c.931C>T, p.Arg311Trp; c.944T>G, p.Val315Gly |
zložený heterozygot: delécia celého génu; p.Gly424Glu |
rovnaká ako Pacient 6 (súrodenci) |
homozygot: c.1411C>T, p.Arg471X |
|
Vek v čase diagnostiky ochorenia |
37 mesiacov |
10 mesiacov |
16dní |
2 dni |
3 dni |
2 mesiace |
1 deň |
2 dni |
|
Dĺžka sledovania |
10 mesiacov |
6 mesiacov |
16 rokov |
8 rokov |
20 mesiacov |
9 rokov |
6 rokov |
13 rokov |
|
Výška (cm) / SDS výšky |
87/-2,3 |
75/+0,69 |
50/+1,19 |
42/-1,4 |
38/+0,3 |
55/-1,38 |
52/+1,02 |
46/+0,3 |
|
Hmotnosť (kg) / SDS hmotnosti |
10,3/-1,89 |
7,06/-2,04 |
2,9/+0,41 |
1,64/-1,4 |
1,07/+0,1 |
3,7/-2,25 |
4,1/+1,53 |
2,2/-0,3 |
|
Gestačný týždeň |
37. |
39. |
37. |
34. |
28. |
39. |
40. |
35. |
|
Polyhydramnion |
áno |
nie |
áno |
áno |
áno |
nie |
áno |
áno |
|
Polyúria-polydipsia |
nie |
nie |
áno |
áno |
áno |
nie |
nie |
áno |
|
pH krvi |
7,52 |
7,61 |
7,56 |
7,53 |
7,4 |
7,67 |
7,49 |
7,45 |
|
HCO3 - (mmol/l) |
28 |
35,7 |
29,1 |
39,3 |
26 |
48,7 |
25,1 |
36,8 |
|
S-Na (mmol/l) |
137 |
131 |
136 |
131 |
117 |
123 |
152 |
132 |
|
S-K (mmol/l) |
3,01 |
2,67 |
2,67 |
2,7 |
10,4 |
2,5 |
6,4 |
2,8 |
|
S-Cl (mmol/l) |
95 |
83 |
93 |
90 |
78 |
82 |
113 |
95 |
|
S-Ca (mmol/l) |
2,62 |
2,33 |
2,57 |
2,67 |
3,62 |
2,71 |
2,48 |
2,73 |
|
S-Mg (mmol/l) |
0,93 |
0,91 |
0,89 |
1,04 |
0,96 |
0,69 |
0,95 |
0,84 |
|
eGFR (ml/min/1,73m2) |
155 |
164,3 |
113 |
75,6 |
160,2 |
120,2 |
125 |
97,8 |
|
Renín (ng/l) |
>329.9 |
>329.9 |
>341 |
>340 |
>340 |
>330 |
>330 |
1434 |
|
Aldosterón (ng/l) |
405,8 |
647 |
407,1 |
>1312 |
>1312 |
1218 |
621 |
4747 |
|
U-Ca/kreat (mmol/mmol) |
2,63 |
1,73 |
2,72 |
4,72 |
13,34 |
1,59 |
2,15 |
1,25 |
|
Nefrokalcinóza |
áno |
nie |
áno |
áno |
áno |
nie |
nie |
nie |
|
Liečba NSAID |
áno |
nie |
áno |
áno |
áno |
áno |
áno |
áno |
|
Suplementácia sodíka |
nie |
áno |
nie |
nie |
nie |
nie |
nie |
nie |
|
Liečba mineralokortikoidným antagonistom |
áno |
áno |
áno |
áno |
nie |
áno |
áno |
áno |
Materiál a metódy
V súbore 8 detských pacientov z Českej republiky (5 pacienti) a Slovenska (3 pacienti) s geneticky potvrdeným Bartterovým syndrómom sme retrospektívne analyzovali klinické prejavy, laboratórne údaje a výsledky molekulovo-genetického vyšetrenia (tab. 2). Súbor tvoria pacienti s Bartterovým syndrómom dispenzarizovaní na pracoviskách autorov. Porovnali sme vstupné hodnoty pri manifestácii ochorenia a pri poslednej ambulantnej kontrole v kontexte aktuálnej liečby. Údaje o hmotnosti a výške sme vyjadrili ako skóre smerodajnej odchýlky podľa noriem Svetovej zdravotníckej organizácie. Za fyziologické rozmedzia sledovaných laboratórnych parametrov sme považovali sérové koncentrácie draslíka 3,50–5,50 mmol/l, sodíka 135 – 145 mmol/l, chloridov 95–107 mmol/l, horčíka 0,70–1,00 mmol/l, bikarbonátov 22,0–26,0 mmol/l a pH 7,36–7,44. Normokalciúria bola definovaná vzhľadom na vek pacienta.(10) Odhad glomerulárnej filtrácie sme kalkulovali podľa vzorca eGFR = 36.5 × výška (cm) / sérový kreatinín (µmol/l).(11)
Výsledky
Súbor tvorí 8 detských pacientov (P1–P8) vo veku od 1,3 do 16 rokov (priemerný vek 7,6 rokov) s geneticky potvrdeným Bartterovým syndrómom. Zastúpenie ženského a mužského pohlavia je vyrovnané (4 : 4), priemerná dĺžka sledovania pacientov je 7 rokov. Väčšinu súboru (5 pacienti) tvoria pacienti s klasickým Barterrovým syndrómom (BS3) na podklade mutácií génu CLCNKB, ktorých priemerný vek v čase prvého záchytu ochorenia bol 9,9 mesiacov. Antenatálny Bartterov syndróm je zastúpený tromi pacientami (dvaja s BS1 a jeden s BS2), ktorí boli diagnostikovaní vo včasnom novorodeneckom období. Ostatné typy (BS4 a BS5) nemajú v našom súbore zastúpenie. P6 a P7 sú súrodenci, čomu zodpovedá rovnaký genetický nález a včasná cielená diagnostika u P7.
Polyhydramnion bol prítomný u 3 pacientov s klasickým BS a u všetkých pacientov s antenatálnou formou ochorenia. Predčasne narodení boli 5 pacienti, no u dvoch chorých s klasickou formou ochorenia išlo len o ľahkú nezrelosť, kým pacienti s antenatálnym BS vykazovali strednú až ťažkú nezrelosť. Polyúria a polydipsia dominovala u pacientov s antenatálnym BS, avšak iba u jedného pacienta s BS3. Traja pacienti s klasickým BS boli v úvode odoslaní pediatrom na širšiu diferenciálnu diagnostiku pre neprospievanie alebo poruchu rastu. Po cielenej diagnostike a adekvátnej substitučnej liečbe sa u každého výrazne zlepšili antropometrické ukazovatele. V laboratórnych parametroch pri prvej manifestácii sme potvrdili takmer u všetkých pacientov typický nález hypokaliemickej hypochloremickej metabolickej alkalózy. Výnimku tvorí P5 s hyperkaliémiou vo veku 3 dní, čo však zodpovedá typu BS2, ktorý je charakterizovaný tranzitórnou novorodeneckou hyperkaliémiou. Za pravdepodobnú príčinu sa pokladá nezrelosť Na+K+ATPázy ako aj fakt, že dysfunkčný draslíkový kanál ROMK je exprimovaný aj v zberných kanálikoch, kde sa spolupodieľa na exkrécii draslíka.(12) Spoločným laboratórnym rysom súboru bola hyponatriémia, respektíve natriémia tesne nad spodnou hranicou referenčných hodnôt. Korešpondujúce laboratórne zmeny v čase prvého vyšetrenia nenachádzame u P7, zrejme vplyvom skorého postnatálneho obdobia, no v ďalšom priebehu sa aj u tohto pacienta rozvinul charakteristický laboratórny fenotyp. Všetci pacienti v súbore mali normokalciémiu a normomagneziémiu, pričom iba P3 vyžaduje dlhodobú substitučnú liečbu magnéziom. Pokles renálnej funkcie zodpovedajúcej štádiu G2 podľa klasifikácie KDIGO sme zaznamenali iba u P4 s BS1. Hyperkalciúriu pri poslednom vyšetrení mali všetci pacienti s výnimkou P3, najvýraznejšie však dominuje u chorých s antenatálnym BS (P4 a P5). Nefrokalcinózu majú všetci pacienti s antenatálnym BS, no iba jedna pacientka (P1) s klasickým BS. Je zaujímavé, že nefrokalcinóza preukázaná v čase diagnostiky u pacientky P3 na pravidelnej liečbe postupne regredovala.
Všetci pacienti užívajú pravidelnú substitúciu draslíka vo forme KCl (priemerná dávka 2,88 mmol K+ / kg / deň). Paradoxne, napriek najvýraznejšej klinickej manifestácii vyžadujú pacienti s antenatálnym BS najnižšie substitučné dávky draslíka. Trvalú substitúciu NaCl vyžaduje iba P2. V čase poslednej kontroly užíva 5 pacientov indometacín (priemerná dávka 0,92 mg/kg/deň). P3 a P8 boli dlhodobo (14 a 6 rokov) liečení indometacínom bez komplikácií. Nesteroidné antiflogistiká neboli doposiaľ indikované iba u P2. S výnimkou P5 sú všetci pacienti dlhodobo na liečbe spironolaktónom (priemerná dávka 0,71 mg/kg/deň). Na symptomatickej terapii majú všetci pacienti vyrovnané vnútorné prostredie, udržuje sa normonatriémia (priemer 138 mmol/l), normo - prípadne ľahká hypokaliémia a hypochlorémia (priemerne 3,6 mmol K+/l a 98 mmol Cl-/l). Priemerné hodnoty pH a bikarbonátov v čase posledného vyšetrenia dosahujú 7,46 a 25,7 mmol HCO3-/l, čo je v porovnaní s iniciálnymi priemernými hodnotami (pH 7,53 a 33,6 mmol HCO3-/l) výrazné zlepšenie.
Diskusia
V retrospektívnej analýze prvej česko-slovenskej kohorty 8 detí s geneticky potvrdeným Bartterovým syndrómom uvádzame komplexné klinické a laboratórne charakteristiky ochorenia. U všetkých pacientov sme potvrdili laboratórny fenotyp hypokaliemickej hypochloremickej metabolickej alkalózy a hyperrenínového hyperaldosteronizmu pri normálnom systémovom krvnom tlaku. Najčastejšie sme zistili klasický BS (62,5%), výskyt polyhydramnionu (75%) a prematurity (62,5%) korešponduje s výsledkami štúdií z Veľkej Británie, Turecka či Južnej Kórey.(13–15) Skorší záchyt ochorenia u našich pacientov s BS1 a BS2 v porovnaní s klasickým typom BS opisujú aj iní autori.(14,16,17) Na druhej strane, Seys a kol. vo veľkej kohorte 115 pacientov s genetickým BS3 až v takmer 30% prípadov pozorovali antenatálnu, prípadne neonatálnu manifestáciu, čo poukazuje na výraznú variabilitu fenotypu ochorenia.(18) Viaceré doteraz publikované štúdie nepotvrdili genotypovo-fenotypovú koreláciu, pričom je zaujímavé, že aj pacienti s rovnakými mutáciami majú odlišný klinický fenotyp.(14,19,20) Príčinou môžu byť modifikujúce gény, faktory prostredia či epigenetické mechanizmy, úlohu môžu zohrávať aj etnické faktory.
U detí s antenatálnou formou ochorenia výraznejšie dominujú klinické prejavy ako polyhydramion, prematurita, polyúria a polydipsia, kým klasická forma ochorenia sa u našich chorých najčastejšie manifestovala neprospievaním či poruchou rastu (60% pacientov s BS3). Symptomatická liečba s úpravou vnútorného prostredia viedla k zlepšeniu sledovaných antropometrických parametrov. V súlade s našimi pozorovaniami aj recentná turecká štúdia preukázala signifikatne vyšší výskyt prematurity, polyúrie a polydipsie u pacientov s antenatálnym BS, a neprospievanie ako dominantný klinický prejav u pacientov s klasickým BS.(14)
Nefrokalcinóza je častá pri BS1 a BS2,(21) čo sme dokumentovali u všetkých troch pacientov s antenatálnym BS. Väčšina autorov uvádza nízky výskyt nefrokalcinózy pri klasickom BS, no z tohto konceptu sa vymykajú dáta kórejskej kohorty s incidenciou nefrokalcinózy v čase diagnostiky až v 21% prípadov BS3.(15) Treba však uviesť, že autori počas longitudinálneho sledovania zaznamenali regresiu nefrokalcinózy takmer u polovice chorých, čo sme pozorovali aj u P3 v našom súbore.
K rozvoju chronickej obličkovej choroby (CKD) pri Bartterovom syndróme prispievajú viaceré faktory (prematurita, opakované ataky dehydratácie, chronické užívanie nesteroidných antiflogistík, nefrokalcinóza). Do akej miery sa na chronickom poškodení obličiek podieľa príslušná génová porucha, nie je celkom jasné.(6) Údaje o CKD u pacientov s Bartterovým syndrómom sa v jednotlivých krajinách sveta významne líšia. Turecko reportuje výskyt CKD (eGFR < 90 ml / min / 1,73m2) u polovice z 21 pacientov s antenatálnym BS (priemerný vek 8,6 rokov) a v 20% prípadov s klasickým BS (priemerný vek 5,1 rokov).(14) Naproti tomu v kórejskej kohorte dospelo 11% chorých z 54 pacientov do CKD (eGFR < 60 ml / min / 1,73 m2) osem rokov od stanovenia diagnózy.(15) Vysoký výskyt CKD reportujú v Británii, kde až 64% pacientov v 15. roku života dosiahlo pokročilé renálne zlyhanie, predominantne pri type BS2.(13) Multivariačná analýza preukázala, že nefrokalcinóza významne predikovala pokles GFR.
Cieľom liečby BS je úprava elektrolytovej dysbalancie a predchádzanie epizódam dehydratácie. Korekciu vnútorného prostredia možno dosiahnuť pravidelnou suplementáciou draslíka a sodíka, účinné sú aj nesteroidné antiflogistiká.(17) Odporúčajú sa u symptomatických pacientov s nedostatočnou odpoveďou na iónovú substitúciu.(6) V našej kohorte sú všetci pacienti na dlhodobej substitúcii chloridom draselným a 7 pacienti užívajú indometacín a spironolaktón. K liečbe antagonistami aldosterónu u detí treba pristupovať opatrne. Pre riziko kritickej hypovolémie sa rutinne neodporúčajú a indikované by mali byť iba v prípade, že kombináciou substitučnej liečby a nesteroidných antiflogistík nie je dosiahnutá kompenzácia ťažkej hypokaliémie.(1)
Podľa našich skúseností je liečba spironolaktónom bezpečná, v priebehu viacročného sledovania sme u žiadneho dieťaťa nezaznamenali epizódu klinicky závažnej hypovolémie. Pacienti liečení diuretikami by však mali byť v dobe obmedzeného príjmu čí zvýšených strát tekutín a elektrolytov (napr. pri akútnej gastroenteritíde) starostlivo monitorovaní a v prípade potreby včasne odoslaní k parenterálnej rehydratácii. Optimálna dávka kália nie je stanovená a môže sa meniť s vekom dieťaťa. Britskí autori preukázali inverznú koreláciu medzi dávkou a vekom dieťaťa s maximálnou potrebou suplementácie v 1. roku života (6,6 mmol/kg/deň resp. 1,5 mmol/kg/d v 15. roku).(13) Substitúciu sodíka vyžaduje iba najmladší pacient zo súboru. Staršie deti si vedia zvýšené straty sodíka hradiť prisoľovaním stravy, v čom ich nie je nutné obmedzovať. Efektívnou kombinovanou symptomatickou liečbou sme u našich pacientov dosiahli dobrú laboratórnu kompenzáciu ochorenia aj pri nižších dávkach indometacínu (odporúčané 1–4 mg indometacínu / kg / deň v 3–4 dávkach).(1)
Limitáciou našej štúdie je relatívne malý počet pacientov a retrospektívny zber dát. Na druhej strane treba vyzdvihnúť, že ide o prvú česko-slovenskú kohortu s najvyšším počtom detí s geneticky potvrdeným Barrterovým syndrómom, u ktorých sme zdokumentovali klinické, laboratórne a genetické charakteristiky. Vytvorenie medzinárodných registrov s väčším počtom pacientov môže významne prispieť k zvýšeniu kvality života chorých s Bartterovým syndrómom. |
Sources
1. Konrad M, Nijenhuis T, Ariceta G, et al. Diagnosis and management of Bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int 2021; 99(2): 324–335.
2. Ji W, Foo JN, O’Roak BJ, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet 2008; 40(5): 592–599.
3. Schnermann J. The juxtaglomerular apparatus: from anatomical peculiarity to physiological relevance. JASN 2003; 14(6): 1681–1694.
4. Ohlsson A, Sieck U, Cumming W, et al. A variant of Bartter’s syndrome.: Barter’s syndrome associated with hydramnios, prematurity, hypercalciuria and nephrocalcinosis. Acta Paediatr 1984; 73(6): 868–874.
5. Stein JH. The pathogenetic spectrum of Bartter’s syndrome. Kidney Int 1985; 28(1): 85–93.
6. Konrad M. Bartter and Gitelman syndromes in children: Clinical manifestations, diagnosis, and management. UpToDate 2021.
7. Laghmani K, Beck BB, Yang SS, et al. Polyhydramnios, transient antenatal Bartter’s syndrome, and MAGED2 mutations. N Engl J Med 2016; 374(19): 1853–1863.
8. Schlingmann KP, Konrad M, Jeck N, et al. Salt wasting and deafness resulting from mutations in two chloride channels. N Engl J Med 2004; 350(13): 1314–1319. .
9. Nuñez-Gonzalez L, Carrera N, Garcia-Gonzalez MA. Molecular basis, diagnostic challenges and therapeutic approaches of Bartter and Gitelman syndromes: a primer for clinicians. IJMS 2021; 22(21): 11414.
10. Seeman T, Janda J. Dětská nefrologie. 2., přepracované a doplněné vydání. Praha: Grada Publishing 2021
11. Schwartz GJ, Munoz A, Schneider MF, et al. New equations to estimate GFR in children with CKD. J Am Soc Nephrol 2009; 20 : 629–637.
12. Fretzayas A, Gole E, Attilakos A, et al. Expanding the spectrum of genetic mutations in antenatal Bartter syndrome type II. Pediatr Int 2013; 55(3): 371–373.
13. Kaur A, Webb NJ, Shenoy M, Hulton SA. Bartter syndrome, 15-year experience in the United Kingdom. J Rare Dis Diag Ther 2018; 4(1).
14. Güven S, Gökçe İ, Alavanda C, et al. Phenotypic and genotypic characteristics of children with Bartter syndrome. Turk J Pediatr 2022; 64(5): 825–838.
15. Choi N, Kim SH, Bae EH, et al. Long-term outcome of Bartter syndrome in 54 patients: A multicenter study in Korea. Front Med (Lausanne) 2023; 10 : 1099840.
16. Han Y, Lin Y, Sun Q, et al. Mutation spectrum of Chinese patients with Bartter syndrome. Oncotarget 2017; 8(60): 101614–101622.
17. Puricelli E, Bettinelli A, Borsa N, et al. Long-term follow-up of patients with Bartter syndrome type I and II. Nephrol Dialysis Transplant 2010; 25(9): 2976–2981.
18. Seys E, Andrini O, Keck M, et al. Clinical and genetic spectrum of Bartter syndrome type 3. JASN 2017; 28(8): 2540–2552.
19. García Castaño A, Pérez De Nanclares G, Madariaga L, et al. Poor phenotype-genotype association in a large series of patients with type III Bartter syndrome. PLoS ONE 2017; 12(3): e0173581.
20. Konrad M, Vollmer M, Lemmink HH, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol 2000; 11(8): 1449–1459.
21. Mrad FCC, Soares SBM, De Menezes Silva LAW, et al. Bartter’s syndrome: clinical findings, genetic causes and therapeutic approach. World J Pediatr 2021; 17(1): 31–39.
Korešpondenčná adresa:
MUDr. Daniel Csomó
Detská klinika LF UK a NÚDCH
Limbová 1
833 40 Bratislava
daniel.csomo@nudch.eu
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2024 Issue 2
Most read in this issue
- Pathophysiology in paediatrics: What laboratory parameters are suitable for nutrition status assessment?
- Vitamin D and immunity
- Cardiovascular and metabolic complications of childhood obesity