
Dystrofinopatie
Authors:
P. Balážová; K. Viestová; Dystrofinopatie M. Kolníková
Authors‘ workplace:
Klinika detskej neurológie, Lekárska fakulta Univerzity Komenského Národný ústav detských chorôb, Bratislava
Published in:
Čes-slov Pediat 2022; 77 (4): 198-205.
Category:
Comprehensive Report
doi:
https://doi.org/10.55095/CSPediatrie2022/032
Overview
Dystrofinopatie predstavujú hereditárne podmienené neuromuskulárne ochorenia zo skupiny svalových dystrofií charakterizované úplnou absenciou alebo zníženou funkciou dystrofínového proteínu. Medzi dystrofinopatie radíme predovšetkým Duchennovu svalovú dystrofiu (DMD), Beckerovu svalovú dystrofiu (BMD) a DMD-asociovanú dilatačnú kardiomyopatiu (DCM). Vzhľadom na incidenciu ochorenia v populácii patria dystrofinopatie medzi najčastejšie neuromuskulárne ochorenia detského veku. V klinickom obraze sa typicky vyskytuje oneskorený motorický vývoj, progresívna svalová slabosť, pseudohypertrofie v oblasti lýtok a Gowersov príznak. Na základe klinických symptómov a pomocných vyšetrení (nález zvýšenej hodnoty kreatínkinázy) je následne diagnóza stanovená molekulárno - genetickým vyšetrením. V terapii je štandardne odporúčaná kortikosteroidná liečba pri súčasnom multidisciplinárnom manažmente pacienta v špecializovaných centrách pre neuromuskulárne ochorenia.
Klíčová slova:
Duchennova svalová dystrofia – Beckerova svalová dystrofia – dystrofinopatie
Úvod
Dystrofinopatie zaraďujeme medzi hereditárne podmienené neuromuskulárne ochorenia zo skupiny svalových dystrofií charakterizované úplnou absenciou alebo zníženou funkciou dystrofínového proteínu. Pojem dystrofinopatie zastrešuje spektrum ochorení, medzi ktoré radíme predovšetkým Duchennovu svalovú dystrofiu (DMD), Beckerovu svalovú dystrofiu (BMD) a DMD-asociovanú dilatačnú kardiomyopatiu (DCM). Dystrofinopatie sa v populácii vyskytujú s frekvenciou 1 : 5000 novorodencov mužského pohlavia, čím sa ochorenie zaraďuje medzi najčastejšie neuromuskulárne ochorenia detského veku.(2) Globálna prevalencia DMD vrátane novorodencov a najstarších žijúcich pacientov sa pohybuje v rozmedzí 0,9 až 16,8 prípadov na 100 000 mužov.(3)
Prvé sporadické prípady pravdepodobne Duchennovej svalovej dystrofie boli popisované už v prvej polovici 19. storočia. V roku 1836 talianski lekári Gaetano Conte (1798–1858) a L. Gioja popísali v Annali Clinici dell’Ospedale degli Incurabili di Napoli klinický obraz dvoch bratov s progresívnou svalovou slabosťou, pseudohypertrofiami lýtok a kontraktúrami, ktoré vznikali v priebehu ochorenia. Francúzsky neurológ Guillaume Benjamin Amand Duchenne de Boulogne v roku 1861 prezentoval svoj prvý prípad DMD pacienta, pričom ochorenie označil ako „paraplegia cerebrale, congenitale, hypertrophique“. Neskôr vo svojich prácach navrhol nový názov ochorenia – tzv. pseudohypertrofická paralýza. Duchenne nebol síce prvým autorom popisujúci fenotyp ochorenia, bol však prvým, ktorý definoval hlavné kľúčové znaky ochorenia. Tiež správne predpokladal, že ochorenie vzniká v dôsledku primárneho postihnutia svalového tkaniva a nie ako následok postihnutia miechy alebo mozgu. Zároveň vyvinul prístroj s názvom „histologická harpúna“, ktorý použil v roku 1865 na bioptický odber svalového tkaniva u 8-ročného DMD pacienta.(4) V roku 1950 bol popísaný miernejší variant ochorenia nemeckým lekárom Petrom Emilom Beckerom.(5)
Etiopatogenéza
Dystrofín predstavuje intracelulárny proteín nachádzajúci sa v svalových vláknach ako dôležitá súčasť tzv. dystrofín - -asociovaného glykoproteínového komplexu (DAGC). Predpokladá sa, že hlavnou funkciou dystrofínu je zabezpečenie stabilizácie membrány svalového vlákna počas svalovej kontrakcie. Umožňuje to priama väzba dystrofínu s intracelulárnymi a transmembránovými proteínmi a nepriama väzba s proteínmi extracelulárnej matrix.
V rámci dystrofínu rozoznávame štyri dómeny (obr. 1):

• N-terminálny koniec (actin-binding domain 1 – ABD1) – viaže sa s kontraktilným proteínom aktínom;
• centrálna tyčinková doména (central rod domain) – úsek je svojím zložením podobný proteínu nazvanému spektrín a zabezpečuje flexibilitu počas svalovej kontrakcie;
• doména bohatá na cysteín (cystein-rich domain) – priamo sa viaže s β-dystroglykánmi, ktoré sa ďalej spájajú s α-dystroglykánmi, sarkoglykánmi a sarcospanom. Následne je cez molekulu laminínu-2 sprostredkované spojenie α-dystroglykánov s extracelulárnou matrix;
• C-koniec (carboxy-terminus) – sa viaže s viacerými intracelulárnymi proteínmi svalového vlákna (syntrofíny a dystrobrevín), ktoré súbežne umožňujú naviazanie enzýmu syntázy oxidu dusnatého (nNOS) s funkciou modulátora cievneho tonusu.(6)
Gén DMD kódujúci proteín dystrofín je lokalizovaný na krátkom ramienku chromozómu X v oblasti Xp21.1 a je zložený zo 79 exónov a 78 intrónov.(7) DMD gén zároveň obsahuje najmenej 7 nezávislých tkanivovo špecifických promótorov, ktoré umožňujú tvorbu rôznych izoforiem proteínu dystrofínu.
Izoformy dystrofínového proteínu (Dp – dystrophin protein variants) boli pomenované na základe miesta ich tvorby a dĺžky v kilodaltonoch. Ich vznik je umožnený použitím promótorov, alternatívneho zostrihu alebo rôznych polyadenylačných signálov.(9) Svalová izoforma dystrofínového proteínu Dp427m, ktorá je exprimovaná v plnej dĺžke a podieľa sa na tvorbe dystrofín-glykoproteínového komplexu, je pomerne známa. V súčasnosti pribúdajú štúdie zamerané na objasnenie funkcií kratších izoforiem dystrofínového proteínu, exprimovaných v ostatných tkanivách ako napr. v CNS alebo retine.
DMD gén je svojou veľkosťou 2,4 Mb považovaný za najväčší ľudský gén, čo ho zároveň do istej miery predisponuje ku vzniku de novo mutácií – až jeden z troch prípadov DMD je spôsobený de novo mutáciou. Najčastejšími mutáciami zapríčiňujúcimi rozvoj ochorenia sú delécie a duplikácie v géne DMD. Delécie a duplikácie sa združujú prevažne v dvoch tzv. hot-spot oblastiach – medzi 45–55 exónom a 2–10 exónom.(10) Okrem uvedených delécií (65%) a duplikácií (6%) exónov u DMD pacientov nachádzame aj zriedkavejšie typy mutácií, ako sú bodové mutácie typu nonsense mutácie (13%) a missense mutácie (4%), malé inzercie a delécie (3%) – percentuálne rozloženie typov mutácií sa podľa rôznych štúdií mierne odlišuje. U časti pacientov s fenotypovým a histopatologickým obrazom dystrofinopatií sa súčasnými metódami molekulárno-genetickej diagnostiky nepodarí potvrdiť mutáciu v DMD géne. V uvedenom prípade možno predpokladať napr. hlboké intrónové mutácie, ktoré spôsobujú začlenenie intrónovej sekvencie do mRNA a vytvorenie tzv. pseudoexónu DMD génu.(11) Začlenenie intrónovej sekvencie do mRNA môže následne narušiť čítací rámec génu alebo vedie k vytvoreniu predčasného stop kodónu.(10)
Vo väčšine prípadov je možné použiť pri korelácii genotypu a fenotypu DMD/BMD tzv. reading frame rule. Uvedené pravidlo vo všeobecnosti sleduje, či daná mutácia zachováva pri syntéze proteínu čítací rámec génu. V prípade porušenia čítacieho rámca (frame-shift mutácia) vzniká predčasné ukončenie translácie v dôsledku prítomnosti predčasného stop kodónu a syntéza skráteného a nefunkčného proteínu. Frame-shift mutácie v géne DMD vedú k rozvoju závažnejšej formy ochorenia. Ak mutácia v DMD géne nenaruší čítací rámec (in-frame mutácia) dochádza k syntéze skráteného proteínu.( 7) Napriek skrátenej dĺžke je vo výslednom proteíne prítomný N-terminálny koniec a C-koniec, zohrávajúci kľúčovú úlohu pri spájaní kontraktilných proteínov a extracelulárnej matrix. Uvedený dystrofín tak parciálne plní svoju fyziologickú funkciu, preto in-frame mutácie bývajú zväčša pozorované u fenotypu BMD (obr. 2).(10)

Klinický obraz
Variabilita klinických príznakov pozorovaných u dystrofinopatií priamo koreluje s množstvom syntetizovaného dystrofínového proteínu. Dystrofinopatie je vo všeobecnosti možné rozdeliť na viaceré fenotypy:
• Duchennova svalová dystrofia (DMD);
• Beckerova svalová dystrofia (BMD);
• DMD-asociovaná dilatačná kardiomyopatia / X-viazaná dilatačná kardiomyopatia (DCM);
• syndróm krampov s myoglobinúriou;
• syndróm s postihnutím intelektu bez svalovej slabosti;
• ženy s fenotypom Duchennovej svalovej dystrofie;
• symptomatické prenášačky.(12)
Duchennova svalová dystrofia
Duchennova svalová dystrofia sa typicky manifestuje v predškolskom veku medzi 3. až 5. rokom života oneskoreným motorickým vývojom alebo príznakmi svalovej slabosti.(13) Ako priemerný vek chlapca s Duchennovou svalovou dystrofiou v čase stanovenia diagnózy sa uvádzajú 4 roky a 10 mesiacov. Napriek pokroku v diagnostike a manažmente DMD pozorujeme významnú latenciu medzi rozvojom klinického obrazu a stanovením diagnózy genetickým vyšetrením.(12)
Medzi prvé pozorované príznaky v dôsledku proximálnej svalovej slabosti patrí kolísavá chôdza, neschopnosť chôdze po schodoch, skákať, bežať alebo postaviť sa. Typicky býva pozorovaný Gowersov príznak (šplhanie sa po stehnách za pomoci rúk), ktorý opísal v roku 1879 neurológ sir William Richard Gowers ako charakteristický príznak pacientov s DMD, vznikajúci v dôsledku svalovej slabosti panvového pletenca a dolných končatín. S ohľadom na súčasné vedomosti Gowersov príznak považujeme za nešpecifický príznak vyskytujúci sa u rôznych klinických jednotiek asociovaných so slabosťou v oblasti panvového pletenca a dolných končatín, zahrňujúcich napr. pletencové svalové dystrofie, spinálnu muskulárnu atrofiu alebo sarkoglykanopatie.(14) Ďalším príznačným fenotypovým znakom je prítomnosť pseudohypertrofií v oblasti lýtok, vznikajúcich v dôsledku depozície tuku a spojivového tkaniva. Zmeny postihujúce zadný kompartment dolných končatín vedú k strate svalov umožňujúcich plantárnu flexiu a následne k rozvoju chôdze po špičkách.(15)
Po úvodnej manifestácii ochorenia u pacientov s DMD pozorujeme tzv. honeymoon phase medzi 3. a 6. rokom života s typickým krátkodobým zlepšením stavu. Nasleduje plató fáza v trvaní niekoľkých mesiacov až jedného roka, ktorá je vystriedaná progresiou ochorenia s lineárnym poklesom svalovej sily smerujúcim k invalidizácii pacienta.(12) Do 13. roku života u väčšiny pacientov s klasickým fenotypom DMD dochádza k strate samostatnej hybnosti. S postupnou progresiou svalovej slabosti a zníženou mobilitou pacienta nastupujú muskuloskeletálne komplikácie ochorenia ako kontraktúry, skolióza a fraktúry v dôsledku osteoporózy ako následok základného ochorenia a súčasnej kortikosteroidnej terapie.(17)
U časti DMD pacientov je prítomné kognitívne postihnutie, neurovývojové poruchy (poruchy autistického spektra, poruchy pozornosti s hyperaktivitou) alebo obsesívno-kompulzívna porucha.(11) Riccoti et al. vo svojej práci pomenovali uvedený súbor porúch vyskytujúcich sa u pacientov s Duchennovou svalovou dystrofiou ako tzv. DMD neuropsychiatrický syndróm.(18) V prípade, že sa neuropsychiatrické príznaky manifestujú pred rozvojom svalovej slabosti, môže dochádzať ku stanoveniu nesprávnej diagnózy a prehliadnutiu diagnózy svalovej dystrofie. Výsledky štúdii zaoberajúcich sa postihnutím CNS u pacientov s DMD nie sú úplne jednoznačné. Pribúdajú však dôkazy, ktoré spájajú popisované neuropsychiatrické príznaky a kognitívne postihnutie v súvislosti s nedostatkom dystrofínu v mozgu. Ako už bolo uvedené, expresia dystrofínu je tkanivovo špecifická a s použitím promótorov, alternatívneho zostrihu polyadenylačných signálov dochádza k syntéze kratších izoforiem dystrofínového proteínu. Mutácie v DMD géne lokalizované v proximálnej oblasti (exón 1–43) sú asociované s absenciou Dp427p,c,m, pričom mutácie v distálnej oblasti (intrón 44 – exón 79) ovplyvňujú expresiu Dp140 a Dp71/Dp40. Práve distálne mutácie DMD génu sú spájané s kognitívnym postihnutím pozorovaným u pacientov s Duchennovou svalovou dystrofiou. V súvislosti s postihnutím CNS je uvádzaná aj vyššia prevalencia epilepsie (6,3%) medzi DMD pacientami v porovnaní s prevalenciou epilepsie v detskej populácii. Aj v tomto prípade sa v patofyziológii vzniku epilepsie predpokladá význam izoforiem dystrofínu a ich rozdielnej regionálnej a subcelulárnej distribúcie. Štúdia s kohortou 14 DMD pacientov ukázala zvýšené riziko epilepsie (7,5%) u nosičov mutácie v oblasti medzi exónom 44 až 63 DMD génu, avšak ide o štúdiu s malým množstvom participantov.(19)
Hlavnú príčinu morbidity a mortality DMD pacientov predstavujú respiračné komplikácie, ako je slabosť respiračných svalov, atelektázy, recidivujúce respiračné infekcie a respiračné zlyhanie. Progresívny pokles pľúcnych funkcií je pozorovaný od skorého adolescentného veku. Kardiologické postihnutie tvorí závažnú súčasť klinického obrazu DMD. Vzhľadom na zlepšujúci sa manažment respiračných funkcií je pozorovaný nárast úmrtí v dôsledku kardiálneho zlyhania. Manifestáciou kardiálneho postihnutia sú zmeny na EKG, rozvoj kardiomyopatií a arytmií. Napriek závažnému kardiálnemu postihnutiu môžeme zaznamenať mierny až asymptomatický klinický obraz ochorenia v dôsledku zníženej fyzickej aktivity u DMD pacientov.(20)
V dôsledku zlepšovania kvality multidisciplinárneho manažmentu a zavedenia kortikosteroidov sa predlžuje priemerné prežívanie DMD pacientov. Jedna zo štúdii zaoberajúca sa mortalitou DMD pacientov porovnávala priemerné prežívanie DMD pacientov narodených v rokoch 1955–1969 s pacientami narodenými v rokoch 1970 až 1994. Autori pozorovali predĺženie priemerného prežívania DMD pacientov z 25,77 rokov na 40,95 rokov v skupine narodených po roku 1970.(21)
Beckerova svalová dystrofia
Beckerova svalová dystrofia patrí medzi dystrofinopatie s výraznou variabilitou fenotypu. Výskyt Beckerovej svalovej dystrofie v populácii je v porovnaní s DMD zriedkavejší. Medzi jednotlivcami nachádzame klasický fenotyp s pletencovým typom svalovej slabosti, ale aj zriedkavejšie formy ochorenia s klinickým obrazom krampov, myalgiami, myoglobinúriou alebo hyperCKémiou.(22) V klinickom obraze BMD pacientov môže dominovať kognitívne postihnutie, poruchy správania, ADHD alebo poruchy učenia.(23) Určitá variabilita je pozorovaná aj v nástupe samotného ochorenia – prvé príznaky sa objavujú medzi 3. až 20. rokom života, pričom priemerný vek pacienta je 12 rokov. Vzhľadom na zvyčajne miernejší priebeh ochorenia v porovnaní s DMD dochádza k strate samostatnej chôdze po 16. roku života a priemerný vek v čase smrti je okolo 40. roka života. Kardiálne zlyhanie patrí u pacientov s BMD medzi časté príčiny smrti.(12) Z hľadiska kardiálneho postihnutia je úvod ochorenia typický dlhou asymptomatickou periódou, v ktorej môžeme zaznamenať asymptomatické arytmie, nešpecifické zmeny na EKG alebo asymptomatickú hypertrofickú kardiomyopatiu. Je potrebné zdôrazniť, že neexistuje korelácia medzi závažnosťou postihnutia kostrového svalstva a kardiálnym postihnutím. V neskoršom období ochorenie sprevádza rozvoj závažných arytmií, hypertrofickej a dilatačnej kardiomyopatie alebo zlyhanie srdca.(24)
DMD-asociovaná dilatačná kardiomyopatia (X-viazaná dilatačná kardiomyopatia, X-LDCM)
Ochorenie sa najčastejšie manifestuje okolo 10. až 20. roku života progresívnym kongestívnym srdcovým zlyhaním v dôsledku dilatačnej kardiomyopatie. V klinickom obraze sa u časti pacientov popisujú krampy alebo myalgie počas fyzickej aktivity, pričom svalová slabosť s atrofiami svalov nemusí byť konštantne prítomná.(25)
Prenášačky a ženy s fenotypom Duchennovej svalovej dystrofie
Dystrofinopatie patria z hľadiska dedičnosti medzi X-viazané ochorenia, s čím súvisí aj výskyt prenášačstva u pacientov ženského pohlavia. Vo väčšine prípadov ide o asymptomatické prenášačky DMD/BMD, avšak až u 40% prenášačiek DMD/BMD dochádza k rozvoju klinického obrazu (manifesting carriers – MCs).(26) U časti pacientiek je pozorovaný závažnejší priebeh typický pre svalové dystrofie, v ostatných prípadoch sa rozvíja skôr mierna svalová slabosť, opakované pády, chôdza po špičkách, ťažkosti pri vstávaní z podlahy alebo oneskorenie motorického vývoja. Hladina kreatínkinázy môže byť v norme alebo zvýšená – vyšetrenie kreatínkinázy tak predstavuje pomerne senzitívnu metódu určenú na skrínning prenášačiek DMD/BMD.(27) V prípade potvrdenia prenášačstva je odporúčané u pacientov doplniť kardiologické vyšetrenie vo včasnom adolescentnom veku, ktoré by malo zahŕňať echokardiografiu a neinvazívne zobrazovacie vyšetrenie, ako je MR vyšetrenie srdca. V prípade normálneho kardiologického nálezu sa odporúča sledovanie vo frekvencii každých 3–5 rokov.(17)
V zriedkavých prípadoch sa môže Duchennova svalová dystrofia manifestovať aj u žien. Rozvoj fenotypu DMD vzniká ako následok delécie / chromozomálnej prestavby v oblasti Xp21.2, absencie X-chromózomu (napr. Turnerov syndróm), uniparentálnej dizómie, prítomnosti zloženej heterozygotnej mutácie v DMD géne alebo inaktivácie X-chromozómu.( 12)
Diagnostika a manažment pacientov
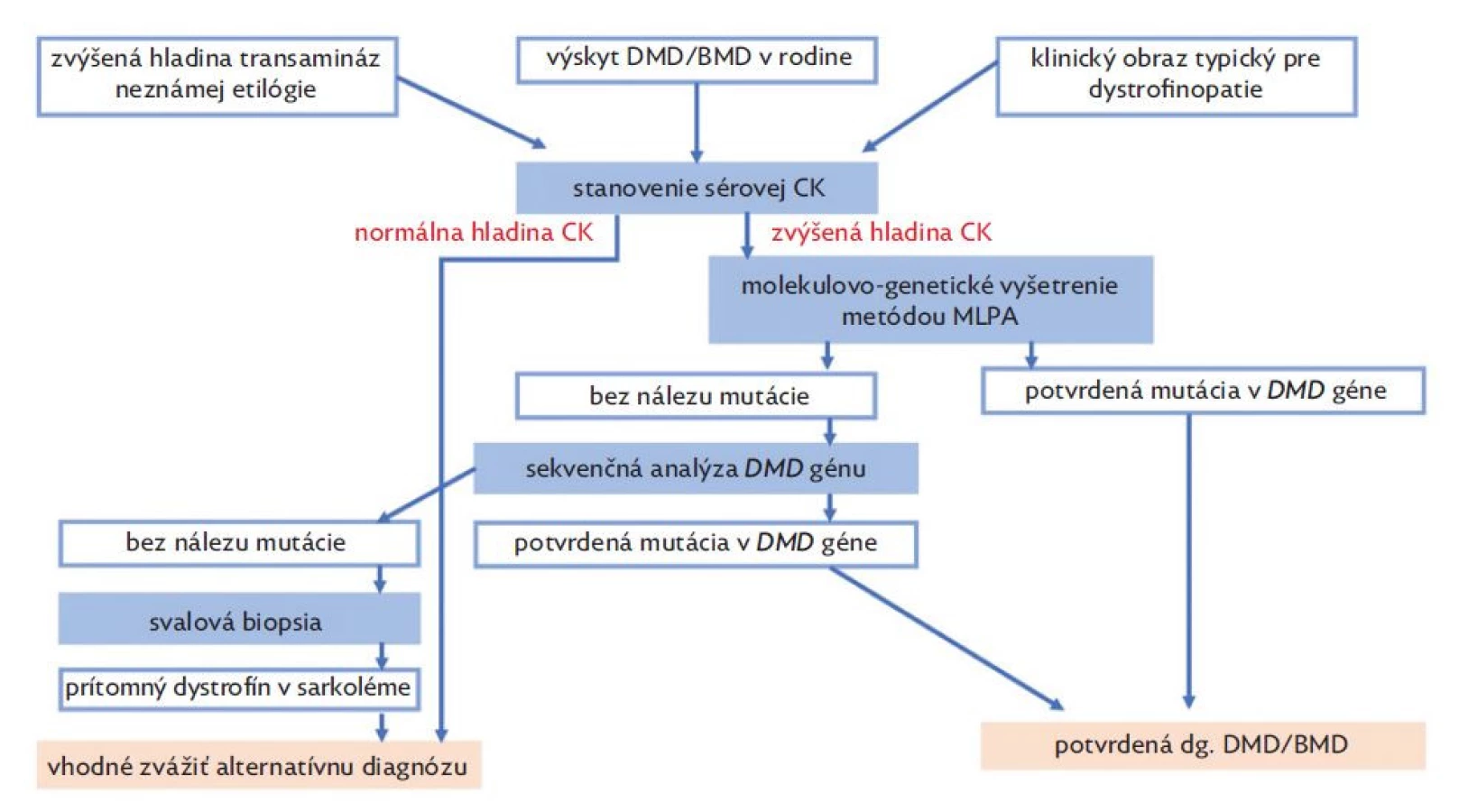
Diagnostika dystrofinopatií je založená na typickom klinickom obraze a výsledkoch pomocných vyšetrení (obr. 3). V laboratórnych vyšetreniach u pacientov nachádzame zvýšenú hladinu sérovej kreatínkinázy (CK), laktátdehydrogenázy (LD) spolu so zvýšenou hladinou transamináz (AST, ALT). U pacientov s DMD zvyčajne nachádzame zvýšenú hladinu CK viac ako 10x normy, u BMD viac ako 5x normy a u prenášačiek môže byť hladina CK v norme alebo až 10x zvýšená.(12) Na základe nálezu elevácie sérových transamináz sú pacienti v mnohých prípadoch odosielaní na gastroenterologické ambulancie s potrebou opakovaných vyšetrení, ktoré zriedkavo končia až biopsiou pečene.(11) Sérové transaminázy (AST, ALT) nie sú špecifické hepatálne enzýmy a k ich uvoľneniu dochádza aj pri poškodení svalových vlákien. Eleváciu transamináz myogénneho pôvodu vždy sprevádza zvýšená hladina CK, pričom hladina GMT a bilirubínu je v referenčnom rozmedzí.(28) Ďalším diagnostickým krokom je v súčasnosti molekulárno-genetické vyšetrenie metódou MLPA za účelom identifikácie delécií a duplikácií v DMD géne. V prípade MLPA negatívneho nálezu sa u pacientov dopĺňa sekvenčná analýza génu, ktorá má vyhľadať zriedkavejšie typy mutácii (bodové mutácie typu nonsense/missense, malé delécie, duplikácie alebo inzercie). Sekvenčnú analýzu možno realizovať Sangerovým sekvenovaním alebo metódou NGS – sekvenovaním novej generácie.(7) V minulosti sa v diagnostike DMD/BMD uplatňovala svalová biopsia, ktorú v súčasnosti nahradili metódy molekulovej genetiky. Pri svalovej biopsii dochádza k stanoveniu prítomnosti dystrofínového proteínu v bioptickej vzorke pomocou imunohistochémie alebo metódou Western blot.(17) V histopatologickom vyšetrení nachádzame nešpecifický obraz nekrózy a regenerácie svalových vlákien s variabilitou ich veľkosti, sprevádzané prítomnosťou vnútorných jadier a endomyziálnou fibrózou.(29)
Rovnako ako svalová biopsia aj elektromyografia už v súčasnosti nepatrí do diagnostického algoritmu DMD/BMD. V EMG nachádzame myogénny nález charakterizovaný prítomnosťou spontánnej aktivity v zmysle fibrilácií a pozitívnych ostrých vĺn, zmenami potenciálov motorických jednotiek (krátkeho trvania s nízkou amplitúdou, často polyfázické potenciály motorických jednotiek) s ich predčasným náborom.(30)
Napriek tomu, že DMD patrí medzi najčastejšie neuromuskulárne ochorenia detského veku, manažment pacientov by mal byť zabezpečený špecializovanými centrami pre neuromuskulárne ochorenia. Špecializované centrá pre neuromuskulárne ochorenia predstavujú ideálny nástroj na poskytovanie multidisciplinárnej starostlivosti o pacienta s ohľadom na aktuálne medzinárodné odporúčania (obr. 4). Pacientom zároveň umožňujú prístup k novým terapeutickým možnostiam. U DMD pacientov je pred výkonom v celkovej anestézii potrebné zohľadniť riziká súvisiace so samotnou anestéziou. Nie je pozorované zvýšené riziko rozvoja malígnej hypertermie v porovnaní s bežnou populáciou. Avšak pri použití depolarizujúcich anestetík (napr. sukcinylcholín) a volatilných plynov (napr. sevofluran, desfluran, halotan) je u dystrofinopatií riziko rozvoja anestéziou indukovanej rhabdomyolýzy a hyperkalémie, preto sa ich použitie neodporúča.(17,31) Očkovanie neuromuskulárnych pacientov nie je vo všeobecnosti spájané so zvýšeným rizikom. V prípade DMD pacientov s plánovanou chronickou kortikosteroidnou liečbou sa odporúča ukončenie vakcinácie podľa očkovacieho kalendára pred začatím samotnej liečby. Počas vysokodávkovej kortikosteroidnej liečby je možné očkovanie inaktivovanými vakcínami, avšak pri očkovaní živými vakcínami sa odporúča prerušiť liečbu na dobu aspoň 1 mesiaca. Súčasne sa odporúča pred začatím kortikosteoidnej liečby očkovanie proti VZV, ak nie je dôkaz o prekonaní ochorenia.(32)

Terapia DMD/BMD pacientov
Kortikosteroidná liečba
Použitie kortikosteroidov za účelom využitia ich protizápalového účinku je vo všeobecnosti považované za zlatý štandard v liečbe DMD pacientov. Medzi benefity dlhodobej kortikosteroidnej liečby zaraďujeme neskoršiu stratu samostatnej chôdze, zachovanie respiračných funkcií a motoriky na horných končatinách. V liečbe sa aktuálne využíva prednizón (v dávke 0,75 mg/kg/deň) a deflazacort (v dávke 0,9 mg/kg/deň), pričom štúdie potvrdili u oboch preparátov efekt na zvýšenie svalovej sily v porovnaní s placebom. Za účelom dosiahnutia maximálneho benefitu je dôležité načasovanie liečby – terapiu sa odporúča indikovať v čase pred signifikantným poklesom motorických schopností pacienta.( 17) Okrem uvedeného prednizónu a deflazacortu prebiehajú štúdie u DMD pacientov s kandidátnym syntetickým steroidom – vamorolonom.(33)
Terapeutické možnosti ovplyvnenia expresie dystrofínu
Posledné desaťročie prináša významné úspechy v oblasti génovej liečby, čím výrazne mení pohľad na terapiu hereditárne podmienených neuromuskulárnych ochorení. Génová terapia otvorila rôzne možnosti ovplyvnenia syntézy dystrofínového proteínu s cieľom obnovenia jeho tvorby a následného zmiernenia fenotypu ochorenia. V prípade potvrdenia nonsense mutácie DMD génu u časti pacientov možno využiť tzv. nonsense supresiu, ktorá bráni vzniku predčasného stop kodónu a následne ukončeniu translácie so vznikom skráteného nefunkčného proteínu. Ataluren (predtým nazývaný tiež PTC124) predstavuje perorálne podávané liečivo zo skupiny aminoglykozidov. Od roku 2014 je schválený Food and Drug Administration (FDA) a European Medicines Agency (EMA) na liečbu DMD pacientov s nonsense mutáciou, pričom uvedenú mutáciu možno potvrdiť u približne 13% pacientov. Metaanalýza dvoch placebom kontrolovaných štúdii s atalurenom v trvaní 48 týždňov ukázala spomalenie progresie ochorenia, pričom signifikantný benefit bol zaznamenaný v skupine pacientov s dosiahnutým výsledkom 6-minútového testu chôdze (6MWD) v rozmedzí 300 až 400 metrov. U pacientov s miernejším fenotypom (6MWD > 400 m) nebolo pozorované významné zlepšenie – pravdepodobne v dôsledku nedostatočnej dĺžky štúdie.(34)
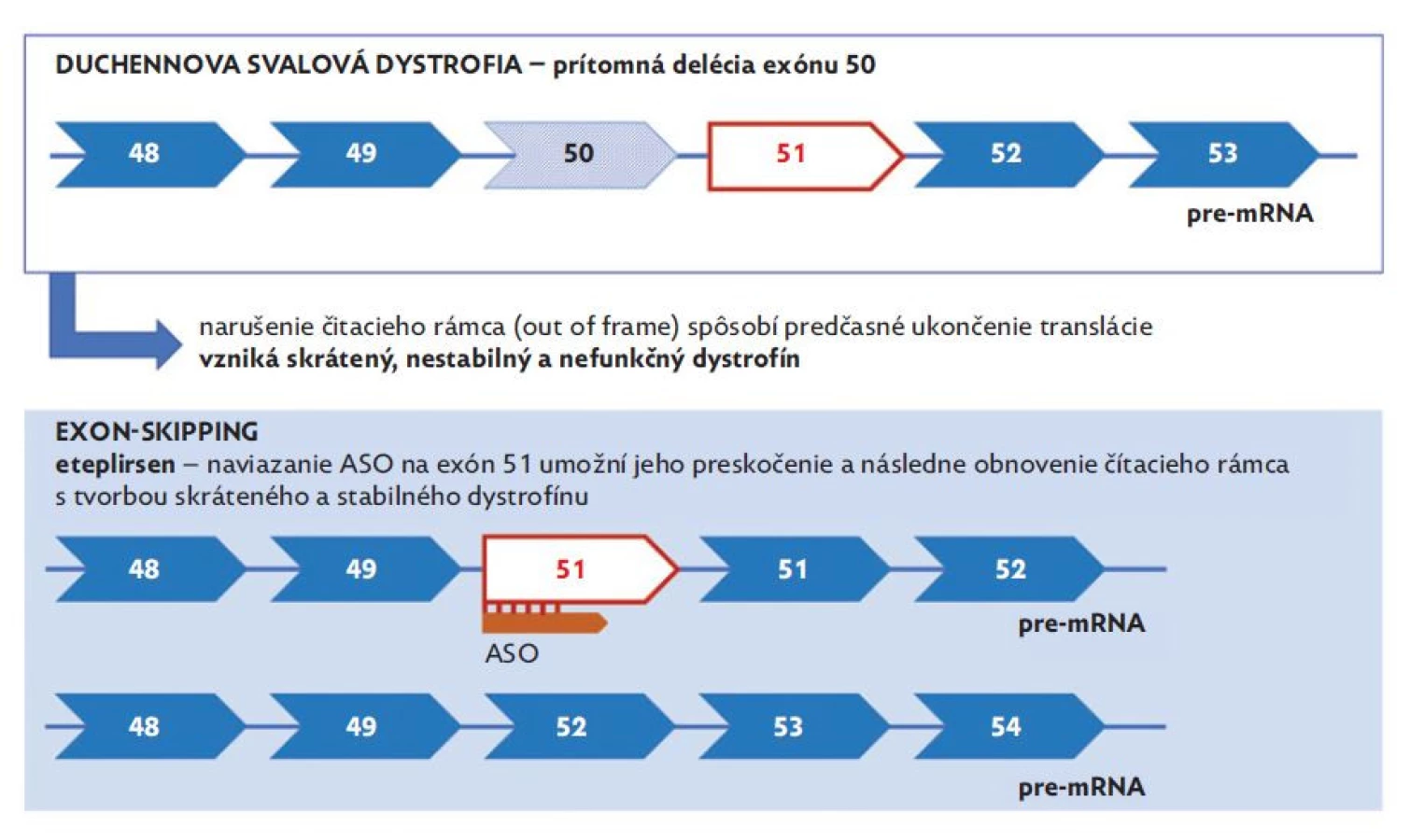
Ďalšiu potenciálnu možnosť ovplyvnenia syntézy dystrofínu predstavuje využitie antisense oligonukleotidov (ASO), ktoré umožňujú moduláciu expresie proteínu pomocou krátkych DNA komplementárnych oligonukleotidov. Napriek tomu, že potenciál ASO je v medicíne známy už dlho, v priebehu rokov bolo potrebné dosiahnuť ich vyhovujúcu stabilitu (tj. odolnosť voči pôsobeniu nukleáz), afinitu, dostatočný prienik do buniek a efektivitu.(35) Aktuálne sa na Slovensku uvedený typ génovej liečby využíva v terapii iného neuromuskulárneho ochorenia detského veku – spinálnej muskulárnej atrofie (nusinersen). V prípade DMD v súčasnosti prebiehajú viaceré klinické štúdie s rôznymi potenciálnymi antisense oligonukleotidmi – eteplirsen (exon 51 skipping), golodirsen (exon 53 skipping), casimersen (exon 45 skipping), viltolarsen (exon 53 skipping), DS-5141b (exon 45 skipping), SRP-5051 (exon 51 skipping) a suvodirsen (exon 51 skipping). Jednotlivé molekuly sa však medzi sebou odlišujú nie len v zložení, ale aj vo svojej efektivite a spektre nežiaducich účinkov.(36) Termín „exon skipping“ v tomto prípade vyjadruje naviazanie antisense oligonukleotidu na špecifický exón s následným preskočením delécie, pričom dochádza k zachovaniu čítacieho rámca a tvorby parciálne funkčného proteínu (obr. 5).(37) Aktuálne je v terapii DMD schválený americkou FDA eteplirsen (Exondys 51) od roku 2016, golodirsen (Vyondys 53) od roku 2019, casimersen (Amondys 45) od roku 2021 a viltolarsen (Vitepso) od roku 2020, pričom ani jeden z liekov nie je registrovaný v EÚ.
Možnosť jednoduchej opravy poškodeného génu u hereditárne podmienených ochorení bola dlho považovaná skôr za fantáziu. Napriek tomu nám 21. storočie ponúka génovú terapiu umožňujúcu náhradu alebo modifikáciu génov pomocou vektorov ako nový terapeutický nástroj v liečbe neuromuskulárnych ochorení. Za účelom transferu génov možno využiť adeno-asociované vírusové vektory (AAV – gene replacement therapy), ktoré sú charakteristické vysokou schopnosťou infikovať rôzne cieľové tkanivá bez potreby integrácie bunkového genómu.(38) Vzhľadom na veľkosť dystrofínového génu, ktorá presahovala možnosti AAV, bol navrhnutý tzv. mini - a mikrodystrofín. Aktuálne prebiehajúce štúdie s využitím systémovej AAV mikrodystrofín génovej liečby demonštrujú zlepšenie motorických funkcií pacienta pri súčasnej zvýšenej expresii mikrodystrofínu v bioptických vzorkách. Medzi nežiadúcimi účinkami sú uvádzané trombocytopénie, akútne obličkové zlyhanie, zvýšenie GMT, atypický hemolyticko-uremický syndróm alebo vracanie.(36)
Ďalšie potenciálne terapeutické ciele
V terapii DMD/BMD majú svoje miesto aj liečivá, ktorých primárny účinok nie je zameraný na ovplyvnenie expresie dystrofínu. Medzi nádejnú terapeutickú skupinu patria inhibítory/ blokátory myostatínu (napr. intravenózna monoklonálna protilátka domagrozumab, solubilný ActRIIB ligand alebo taldefgropbep-α), ktoré však nepreukázali svoju efektivitu v doteraz realizovaných štúdiách.(39) Medzi ďalšie zvažované liečiva patria napr. antioxidanty (napr. idebenon), selektívne modulátory androgénových receptorov (SMARs) alebo inhibítory nukleárneho faktoru – kappa B.
Záver
Dystrofinopatie patria medzi najčastejšie nervosvalové ochorenia. S rýchlo pribúdajúcimi terapeutickými možnosťami je veľmi dôležité správne a rýchle stanovenie diagnózy, čo otvára možnosti nie len nových terapeutických stratégií, ale aj otázku potreby novorodeneckého skrínningu DMD. V niektorých štátoch prebehli pilotné programy založené na stanovení hladiny sérovej kreatínkinázy v rámci novorodeneckého skrínningu. Okrem úspešne diagnostikovaných DMD pacientov súčasne odhalili aj možné nedostatky skrínningu, akými sú falošne pozitívne nálezy hyperCKémie v novorodeneckom veku. Vyšetrenie kreatínkinázy však vidíme ako veľmi užitočné a odporúčame ho zaradiť ako vyšetrenie u všetkých chlapcov vo veku 4–6 mesiacov a u tiež u detí s oneskorením PMV a hypotóniou.
Korešpondenčná adresa:
MUDr. Patrícia Balážová
Klinika detskej neurológie LF UK a NÚDCH
Limbová 1
833 40 Bratislava
Sources
1. Thangarajh M. The Dystrophinopathies. Continuum (Minneap Minn) 2019; 25(6): 1619–1639.
2. Duan D, Goemans N, Takeda S, et al. Duchenne muscular dystrophy. Nat Rev Dis Primers 2021; 7(1): 1–19.
3. Crisafulli S, Sultana J, Fontana A, et al. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta–analysis. Orphanet J Rare Dis 2020; 15(1): 141.
4. Tyler KL. Origins and early descriptions of “Duchenne muscular dystrophy”. Muscle Nerve 2003; 28(4): 402–422.
5. Mrázová L. Duchennova svalová dystrofie – patogeneze, klinický obraz, diagnostika, aktuální možnosti terapie. Neurológia 2016; 11(1): 13–15.
6. Gieron-Korthals M, Fernandez R. New developments in diagnosis, treatment, and management of Duchenne muscular dystrophy. Adv Pediatr 2020; 67 : 183–196.
7. Petrovič R, Fischerová K, Chandoga J. Duchennova muskulárna dystrofia – diagnostika a štuktúra molekulárnogenetických patológií na Slovensku. Neurológia. 15(1): 25–28.
8. Doorenweerd N. Combining genetics, neuropsychology and neuroimaging to improve understanding of brain involvement in Duchenne muscular dystrophy – a narrative review. Neuromuscul Disord 2020; 30(6): 437–442.
9. Naidoo M, Anthony K. Dystrophin Dp71 and the Neuropathophysiology of Duchenne Muscular Dystrophy. Mol Neurobiol 2020; 57(3): 1748–1767.
10. Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet 2016; 53(3): 145–151.
11. Flanigan KM. Duchenne and Becker muscular dystrophies. Neurol Clin 2014; 32(3): 671–688, viii.
12. Brandsema JF, Darras BT. Dystrophinopathies. Semin Neurol 2015; 35(4): 369–384.
13. Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health 2015; 51(8): 759–764.
14. Shrestha S, Munakomi S. Gower Sign. In: StatPearls. StatPearls Publishing; 2022. Accessed March 9, 2022. http: //www.ncbi.nlm.nih.gov/books/NBK540973/
15. Torriani M, Townsend E, Thomas BJ, et al. Lower leg muscle involvement in Duchenne muscular dystrophy: an MR imaging and spectroscopy study. Skeletal Radiol 2012; 41(4): 437–445.
16. Bello L, Morgenroth LP, Gordish-Dressman H, et al. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology 2016; 87(4): 401–409.
17. Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018; 17(4): 347–361.
18. Ricotti V, Mandy WPL, Scoto M, et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol 2016; 58(1): 77–84.
19. Pane M, Messina S, Bruno C, et al. Duchenne muscular dystrophy and epilepsy. Neuromuscul Disord 2013; 23(4): 313–315.
20. Shih JA, Folch A, Wong BL. Duchenne muscular dystrophy: the heart of the matter. Curr Heart Fail Rep 2020; 17(3): 57–66.
21. Ryder S, Leadley RM, Armstrong N, et al. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis 2017; 12(1): 79.
22. Yuan R, Yi J, Xie Z, et al. Genotype-phenotype correlation in Becker muscular dystrophy in Chinese patients. J Hum Genet 2018; 63(10): 1041–1048.
23. Ferrero A, Rossi M. Cognitive profile and neuropsychiatric disorders in Becker muscular dystrophy: A systematic review of literature. Neurosci Biobehav Rev 2022; 137 : 104648.
24. Finsterer J, Stöllberger C. Cardiac involvement in Becker muscular dystrophy. Can J Cardiol 2008; 24(10): 786–792.
25. Nakamura A. X-linked dilated cardiomyopathy: a cardiospecific phenotype of dystrophinopathy. Pharmaceuticals (Basel) 2015; 8(2): 303–320.
26. Imbornoni L, Price ET, Andrews J, et al. Diagnostic and clinical characteristics of early–manifesting females with Duchenne or Becker muscular dystrophy. Am J Med Genet A 2014; 164A(11): 2769–2774.
27. Zhong J, Xie Y, Bhandari V, et al. Clinical and genetic characteristics of female dystrophinopathy carriers. Mol Med Rep 2019; 19(4): 3035–3044.
28. Špalek P. HyperCKémia – etiológia, klinický význam a diferenciálna diagnostika. Via practica 2008; 15 : 259–264.
29. Bednařík J. Svalové dystrofie. Neurol Praxi 2004; 3 : 137–141.
30. Paganoni S, Amato A. Electrodiagnostic evaluation of myopathies. Phys Med Rehabil Clin N Am 2013; 24(1): 193–207.
31. van den Bersselaar LR, Riazi S, Snoeck M, et al.; Anaesthesia and Neuromuscular Disorders Working Group. 259th ENMC international workshop: Anaesthesia and neuromuscular disorders 11 December,2020 and 28–29 May 2021. Neuromuscul Disord 2022; 32(1): 86–97.
32. Golli T, Kastrin A, Pokorn M, Rener-Primec Z. Immunosuppression and immunization: Vaccination in pediatric patients with neuromuscular diseases treated with steroids or immune-modulating drugs. Eur J Paediatr Neurol 2021; 35 : 158–164.
33. Mah JK, Clemens PR, Guglieri M, et al. Efficacy and safety of vamorolone in Duchenne muscular dystrophy: a 30-month nonrandomized controlled open–label extension trial. JAMA Netw Open 2022; 5(1): e2144178.
34. Campbell C, Barohn RJ, Bertini E, et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J Comp Eff Res 2020; 9(14): 973–984.
35. Brenner D, Ludolph AC, Weishaupt JH. Gene specific therapies – the next therapeutic milestone in neurology. Neurol Res Pract 2020; 2 : 25.
36. Fortunato F, Rossi R, Falzarano MS, Ferlini A. Innovative therapeutic approaches for Duchenne muscular dystrophy. J Clin Med 2021; 10(4): 820.
37. Shieh PB. Emerging strategies in the treatment of Duchenne muscular dystrophy. Neurotherapeutics 2018; 15(4): 840–848.
38. Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, et al. Current clinical applications of in vivo gene therapy with AAVs. Mol Ther 2021; 29(2): 464 – 488.
39. Rybalka E, Timpani CA, Debruin DA, et al. The failed clinical story of myostatin inhibitors against Duchenne muscular dystrophy: Exploring the biology behind the battle. Cells 2020; 9(12): E2657.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2022 Issue 4
Most read in this issue
- Sepse u dětí
- Diferenciální diagnostika mikroskopické hematurie
- Hypertermie, její příčiny a rizika z pohledu patofyziologa
- Dystrofinopatie