
Kožní nemoci způsobené monoklonálním imunoglobulinem a/nebo volnými lehkými řetězci (monoklonální gamapatie kožního významu)
Authors:
Z. Adam 1; Z. Řehák 2; M. Krejčí 1; I. Boichuk 1; L. Pour 1
Authors‘ workplace:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Oddělení nukleární medicíny, MOÚ Brno
2
Published in:
Klin Onkol 2026; 39(1): 10-30
Category:
Review
doi:
https://doi.org/10.48095/ccko202610
Overview
Východiska: Monoklonální imunoglobuliny (M-Ig) a/nebo volné lehké řetězce (free light chains – FLC) mohou způsobovat velmi pestrá orgánová a tkáňová poškození. Nejčastější jsou různé formy nefropatií, pro něž byl vytvořen termín „monoclonal gammopathy of renal significance“ a byla přijata morfologická klasifikace vytvořená skupinou International Kidney and Monoclonal Gammopathy Research Group. Další velkou skupinu tvoří klinické jednotky, u nichž vznikají mimo jiné také různé formy poškození kůže. Pro ně bylo vytvořeno skupinové označení „monoclonal gammopathy of cutaneous significance“. Etiopatogeneze je sice hematologická, ale příznaky přivádějí pacienty s touto formou poškození monoklonální gamapatií do dermatologických ambulancí. Cíl: Dermatologické publikace z posledních 5 let rozlišují choroby s jasně vyjádřenou etiopatogenetickou souvislostí s monoklonální gamapatií, těch definují celkem 12, a pak uvádějí další choroby, u nichž je v jednotlivých případech etiopatogenetická souvislost méně jasná, tyto choroby mohou mít i další příčiny. Do skupiny kožních chorob s jasně prokázanou souvislostí s M-Ig patří: AL-amyloidóza kůže, kožní makroglobulinóza, změny kůže při POEMS syndromu, skleromyxedém, skleredém, plošné normolipemické xantomy, nekrobiotický xantogranulom, urtikárie při syndromu Schnitzlerové, teleangiektazie při TEMPI syndromu, lokalizovaný erytém při AESOP syndromu, kryoglobulinemické změny kůže, cutis laxa (volná kůže). Z chorob s méně jasnou souvislostí s M-Ig zmiňujeme IgA pemphigus. V textu uvádíme stručné popisy doplněné obrazovou dokumentací jmenovaných chorob, s nimiž jsme se za 35 let setkali. Závěr: V případě, že se dermatolog s uvedenými chorobami setká, měl by vyšetřit M-Ig a FLC a při pozitivním nálezu konzultovat s hematologem možnost cílené léčby.
Klíčová slova:
volné lehké řetězce – monoklonální imunoglobulin – monoklonální gamapatie kožního významu
Úvod
Změny, které probíhají na kožním povrchu, jsou velmi často důsledkem chorob vnitřních orgánů a tkání. Souvislosti kožních změn s vnitřními chorobami velmi dobře vystihuje kniha Kožní změny u interních onemocnění [1]. A také monoklonální gamapatie mohou indukovat určité typy kožních patologií. Jedním z objevitelů skupiny kožních nemocí, které úzce souvisí s monoklonálním imunoglobulinem (M-Ig), byl Dan Lipsker. V roce 2017 vydal publikaci Monoclonal gammopathy of cutaneous significance: review of a relevant concept, která představila novou koncepci pohledu na některé kožní nemoci. Definoval skupinu kožních nemocí, v jejichž etiopatogenezi mají klíčovou úlohu produkty klonální buněk, tvořících M-Ig. Tedy nejen M-Ig, ale v některých případech i další substance tvořené těmito buňkami. Termín „monoclonal gammopathy of cutaneous significance“ byl akceptován mezinárodní vědeckou komunitou a je standardně používán [2]. V roce 2025 představuje skupina kožních nemocí způsobených M-Ig velkou podskupinu nemocí, které patří do vyšší kategorie zvané monoclonal gammopathy of clinical significance (MGCS).
Současná literatura zabývající se tématem monoclonal gammopathy of cutaneous significance neboli kožními změnami etiopatogeneticky souvisejícími s M-Ig rozděluje kožní projevy monoklonálních gamapatií do dvou skupin:
- kožní změny s jasně prokázanou etiopatogenetickou vazbou na M-Ig. Ty vždy vymizí při potlačení monoklonální gamapatie.
- kožní změny nejednoznačné etiopatogenetické souvislosti s M-Ig. Ty mohou mít více etiologických souvislostí, proto jejich reakce na potlačení monoklonální gamapatie není konstantní.
Rozdělení kožních změn dle míry etiopatogenetické souvislosti s M-Ig z pohledu roku 2025 uvádí tab. 1, která vychází z citovaných publikací [3–6]. Toto dělení má zásadní vliv na léčbu.
![Etiopatogenetická souvislost kožních změn s monoklonálním imunoglobulinem z pohledu roku 2023 [2–5].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/3af75657aef4c0315bbfb04394ec65f5.png)
Léčba kožních poruch s prokázanou etiopatogenetickou souvislostí s M-Ig má za cíl zcela potlačit tvorbu M-Ig neboli dosáhnout kompletní remise. Dosažení parciální remise totiž nezastaví proces poškozující tkáně! K dosažení kompletní remise je nutné použít nejúčinnější léčbu, kterou v roce 2025 představují kombinace obsahují monoklonální protilátku proti antigenu plazmatických buněk – CD38 (daratumumab nebo isatuximab). Otevřenou otázkou je, co bude v roce 2026 a v dalších letech považováno za nejúčinnější dostupnou léčbu.
Pokud by se nepodařilo dosáhnout u těchto chorob kompletního vymizení M-Ig, je léčbou druhé linie obvykle udržovací léčba, aplikace preparátů nitrožilních imunoglobulinů (intravenous immunoglobulins – IVIG) v imunomodulačních dávkách (2 g/kg 1× za 28 dní).
Léčba kožních změn nejednoznačně souvisejících s M-Ig je spíše symptomatická.
Text uvádí stručnou charakteristiku jednotlivých klinických jednotek první skupiny, tj. způsobených monoklonální gamapatií. Klinické jednotky zde uvedené mají kromě dermatologických i jiné projevy. Dermatologické projevy jsou však velmi dobře rozpoznatelné, což je spojuje do jedné podskupiny monoclonal gammopathy of cutaneous significance. Změny kůže, u nichž je vhodné vyšetřit přítomnost M-Ig a volných lehkých řetězců (free light chains – FLC), uvádí tab. 2. Jejich etiopatogenezi ilustruje schéma 1.

![Schéma 1. Dělení kožních projevů monoklonální gamapatie dle předpokládané etiopatogeneze [61]. AESOP – adenopathy and extensive skin patch overlying a plasmacytoma, etiopat. – etiopatogeneze, FLC – volné lehké řetězce, M-Ig – monoklonální imunoglobulin, POEMS – polyneuropatie, organomegalie, endokrinopatie, monoklonální gamapatie, kožní změny, sy – syndrom, TEMPI – teleangiektázie, erytrocytóza, monoklonální gamapatie, perinefritická kolekce tekutiny, intrapulmonální arteriovenózní spojka](https://pl-master.mdcdn.cz/media/image/75bef39642790f69bd0686fdb96506f7.png?version=1772165291)
Kožní změny s vysokou pravděpodobností etiopatogenetické souvislosti s M-Ig a/nebo FLC
AL-amyloidóza kůže
AL-amyloidóza je multisystémové onemocnění, míra poškození orgánů se liší tropizmem amyloidotvorných lehkých řetězců, a proto se projevy nemoci u jednotlivých pacientů liší. Kůže bývá velmi často poškozena, proto zde AL-amyloidózu zmiňujeme.
AL-amyloidóza je termín pro ukládání FLC, obvykle typu lambda, ve formě depozit s lineární strukturou v různých orgánech a tkáních. Klonální plazmocyty, které tyto amyloidotvorné lehké řetězce produkují, mohou mít charakter jak nemaligní populace se zastoupením v kostní dřeni < 10 % všech jaderných buněk, tak i charakter myelomové populace se zastoupením ≥ 10 %. V prvním případě se jedná o primární AL-amyloidózu, která je řazena do skupiny MGCS, v druhém případě jde o mnohočetný myelom s projevy AL-amyloidózy. Mezi těmito formami je kontinuální přechod a arbitrární hranicí tvoří uvedené 10% zastoupení klonální plazmocelulární populace v kostní dřeni. Amyloidová depozita jsou diagnostikována u 10–13 % pacientů s mnohočetným myelomem, pokud se po nich pátrá. Podrobné popisy AL-amyloidózy, která potenciálně může poškodit všechny orgány a tkáně, jsou dostupné v recentní české odborné literatuře [7,8].
Zde jenom připomeneme její kožní projevy. AL-amyloidóza způsobuje zvýšenou fragilitu kožních cév. Obvyklým projevem jsou modřiny snadno vznikající při mechanické zátěži kůže anebo při zvýšení žilního tlaku. To je důsledkem amyloidové vaskulopatie. Proto mezi typické projevy AL-amyloidózy patří purpura a ekchymózy. Hematomy se často tvoří v oblasti víček po zvýšení žilního tlaku, např. při zakašlání. Hematomy víček popisuje Lipsker u 16 % pacientů s AL-amyloidózou. Vlivem AL-amyloidózy dochází k atrofii kůže a její značné fragilitě, kožní poranění vznikají velmi snadno při jakékoliv drobné traumatizaci kůže. Vzácně vznikají depozita amyloidu v kůži vytvářející na kožním povrchu voskovité papuly, plaky či noduly. Na krvácivých projevech se může podílet deficit koagulačního faktoru X, který je adsorbován na amyloidová depozita v játrech. Další příčinou krvácivých projevů může být intenzivnější fibrinolýza.
Ke změnám náležejícím k AL-amyloidóze patří i třepící se nehty s narušenou strukturou nehtu [1]. Kožní projevy AL-amyloidózy a také makroglosii ilustruje obr. 1. Depozita AL-amyloidu mohou vznikat vzácně i u Waldenströmovy makroglobulinemie a poškozovat játra, ledviny a další tkáně.

AL-amyloidóza je bohužel u většiny pacientů diagnostikována až při značném poškození tkání. Její počáteční příznaky (únava, dušnost a otoky dolních končetin) jsou často lékaři nesprávně interpretovány jako projevy ischemické nemoci srdeční a takto jsou i léčeny. Teprve až vyšetření albuminu v séru a bílkoviny v moči prokáže nefrotický syndrom a echokardiografie zjistí hypertrofickou kardiomyopatii s možnou příčinou v amyloidových depozitech, může být diagnostika nasměrována správně.
Kožní makroglobulinemie
Termín kožní makroglobulinemie se používá pro nález papulózních morf, obvykle nad extenzory končetin, které obsahují depozita M-Ig typu IgM. Při histologickém vyšetření těchto papul je nalézán amorfní hyalinní materiál a eozinofilní depozita. Tyto projevy jsou označovány termínem kožní makroglobulinemie (cutaneous macroglobulinosis). Zcela výjimečně byly popsány i bulózní změny kůže u pacientů s Waldenströmovou makroglobulinemií s tvorbou subepidermálních bul [9]. S časnou diagnostikou Waldenströmovy makroglobulinemie ubývá případů těchto kožních projevů monoklonální gamapatie typu IgM. Tím si vysvětlujeme, že s popsanými projevy jsme se na našem pracovišti nesetkali.
POEMS syndrom a osteosklerotický myelom
POEMS syndrom je multisystémové onemocnění, které má mimo jiné i kožní projevy viditelné při podrobném prohlédnutí pacienta. POEMS syndrom je akronymem složeným z prvních písmen slov, které pojmenovávají jeho dominantní příznaky: polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, skin changes.
U POEMS syndromu dominují potíže plynoucí z neurologického poškození, na první pohled jsou ale viditelné kožní změny s touto chorobou spojené. Jsou to: nově se tvořící glomeruloidní hemangiomy, hyperpigmentace, hypertrichóza, akrocyanóza a vznik bělavého zabarvení nehtů. Další znaky POEMS syndromu jsou: městnavá papila na očním pozadí a někdy sklerodermoidní změny v obličeji spojené s atrofií kůže. Pacienti s POEMS syndromem si mohou stěžovat na Raynaudův syndrom. Laboratorní vyšetření by mělo odhalit abnormality v hladinách hormonů, včetně změn v oblasti hypofyzárních hormonů, tedy nějakou formu endokrinopatie.
Pro POEMS syndrom je typická zvýšená sérová koncentrace vaskulárního endoteliálního růstového faktoru (vascular endothelial growth factor – VEGF). Ten je zodpovědný za neuropatii a tvorbu hemangiomů. Vyšetření VEGF je důležité pro diferenciální diagnostiku neuropatie u monoklonálních gamapatií. Zvýšená hodnota VEGF u pacienta s monoklonální gamapatií a neuropatií signalizuje vývoj směrem k POEMS syndromu. Proto je hodnota VEGF zohledněna v posledních kritériích tohoto onemocnění. Pro POEMS syndrom je typická osteoskleróza, jak ilustruje obr. 2. POEMS syndrom může být i příčinou sekundární polyglobulie. Diagnóza tohoto onemocnění je někdy stanovena v rámci diferenciální diagnostiky neuropatie. Poslední verzi kritérií z roku 2023 uvádí tab. 3. POEMS syndrom může být někdy asociován s idiopatickou multicentrickou Castlemanovou nemocí. V těchto případech s prokázaným M-Ig se považuje monoklonální gamapatie za příčinu všech těchto změn

![Kritéria POEMS syndromu dle publikace Angely Dispenzieri z roku 2023 [10].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/12c3bdc04cf6df8cb868700b1743ff00.png)
Cílem léčby je potlačení monoklonální gamapatie dostupnými léky, a jak je vidět z citované literatury, uplatní se zde i bispecifické protilátky [10–15].
V práci anglických autorů z roku 2019 byl u 77 pacientů s POEMS syndromem vyšetřen mozek MR zobrazením. Pachymeningeální změny byly detekovány u 70,7 % pacientů! Většinou bylo ale pachymeningeální zesílení asymptomatické. Autoři práce považují MR mozku s průkazem pachymeningitidy za další známku POEMS syndromu, protože u kontrolní skupiny s jinými příčinami polyneuropatie změny mozkových plen nebyly [16–18].
Osteosklerotický myelom
Osteoskleróza je součástí POEMS syndromu, ale může se u gamapatií vyskytnout bez dalších projevů POEMS syndromu. V těchto případech se pro onemocnění používá termín osteosklerotický myelom, ačkoliv nízkým počtem plazmocytů a obvykle i nízkou koncentrací M-Ig nenaplňuje klasická kritéria myelomu. Tyto případy jsou ale ještě vzácnější než POEMS syndrom. Osteosklerotický myelom a POEMS syndrom v případech, kdy jsou ložiska osteosklerózy v kalvě, indukují zduření přilehlých mozkových plen. U jedné naší pacientky s osteosklerotickým ložiskem ve vrcholu očnice vedla reakce mozkových obalů ke krátkodobé poruše zraku [19,20].
Skleromyxedém
Skleromyxedém (scleromyxedema) je vzácná systémová mucinóza charakterizovaná generalizovaným výsevem voskových drobných papul v terénu tuhé indurované kůže a podkoží, která je obvykle asociovaná s monoklonální gamapatií. Pro chorobu je charakteristický vznik depozit mucinu v pojivové tkáni a proliferace fibroblastů. Nemoc vytváří v kůži ložiska, která jsou tuhá, voskového vzhledu, s papulami velkými 2–3 mm v průměru. Postihuje především oblast hlavy a krku, horní část trupu, předloktí a stehna. Na čele bývá přítomno podélné rýhování, což podmiňuje vzhled typu facies leontina. Ztuhnutí kůže způsobuje omezení hybnosti v postižených oblastech. V případech skleromyxedému je monoklonální gamapatie nalézána ve > 80 % případů, obvykle IgG-k, ostatní typy M-Ig jsou vzácnější. Koncentrace monoklonálního gamaglobulinu bývá ve většině případů nižší než 10 g/l. Maligní gamapatie jsou mezi pacienty se skleromyxedémem podstatně vzácnější, objevují se v 10 % případů skleromyxedému. Zcela výjimečné jsou popisy případů této mucinózy provázející jiné maligní choroby a mizící po jejich vyléčení.
Předpokládá se, že produkty klonálních plazmocytů stimulují proliferaci fibroblastů a tvorbu mucinu v nich. V experimentální studii bylo prokázáno, že sérum pacienta (ale nikoliv purifikovaný M-Ig) stimulovalo proliferaci fibroblastů. Důkazem, že etiopatogenetickou příčinou je monoklonální gamapatie, jsou četné popisy případů, u nichž kompletní potlačení tvorby M-Ig (dosažení kompletní hematologické remise) bylo spojeno s vymizením projevů skleromyxedému [21–25].
Mucinóza nepoškozuje jen kůži, ale objevují se i další příznaky této v podstatě systémové nemoci v následujícím pořadí:
- poškození nervového systému;
- poškození pohybového aparátu;
- poškození kardiovaskulárního systému;
- poškození trávicího traktu, plic a ledvin.
Nervový systém je poškozen asi ve 30 % případů. Skleromyxedém může způsobit senzorickou či motorickou neuropatii nebo syndrom karpálního tunelu. Může také poškodit centrální nervový systém, což se projeví jako kognitivní disfunkce, epilepsie, psychózy. Nejzávažnější variantou poškození CNS skleromyxedémem je takzvaný dermatoneurosyndrom. Je to vzácná febrilní neurologická komplikace s „flu-like“ neboli chřipkovými prodromálními stavy, které mohou progredovat přes epileptiformní záchvaty do kómatu a způsobit smrt nemocného [21–24]. Skleromyxedém je choroba, která nejen zhoršuje kvalitu života, ale také jej výrazně zkracuje. V jedné z mála multicentrických studií obsahující 30 pacientů s touto diagnózou byla mortalita 20 % při mediánu sledování 33 měsíců. Pacienti umírali na mimokožní komplikace skleromyxedému: dermatoneurosyndrom, infarkty myokardu, srdeční selhání [21].
Diagnóza se stanoví dle Rongiolettiho [21] na základě vyšetření následujících parametrů:
- zhodnocení dermatologické, přítomnost generalizované papulární a skleroderomiformní erupce;
- histologické vyšetření kůže a podkoží, zda je splněna histologická triáda: depozita mucinu, fibroblastová proliferace a fibróza;
- laboratorní průkaz M-Ig;
- nepřítomnost onemocnění štítné žlázy dle endokrinologického vyšetření.
Léčba je možná potlačením tvorby M-Ig, a pokud se to nepodaří, tak léčba imunomodulační dávkami IVIG. Naše pacienty s morfy skleromyxedému ilustruje obr. 3.

Skleredém
Skleredém je nemoc ze skupiny mucinóz, která je také asociovaná s M-Ig podobně jako skleromyxedém. Klinicky se projevuje prknovitou indurací a ztluštěním kůže. Skleredém postihuje celý kožní povrch. Někdy se této jednotce říká „skleredema adultorum Buschke“. Je to vzácné onemocnění s excesivní tvorbou mucinu, jak výše uvedeno, ale také se sklerotickými změnami. Při histologickém vyšetření se proces liší od skleromyxedému, u kterého dochází ke zvýšené proliferaci fibroblastů. U skleredému se pod intaktní epidermis nachází ztluštělá dermis s rozšířenými prostory mezi kolagenními svazky, ve kterých je uložen mucin. K proliferaci fibroblastů nedochází. Skleredém nejčastěji postihuje trup a ramena. Zmnožení mucinu však nepostihuje pouze kůži, ale i další orgány a tkáně, takže pacienti mají další závažné projevy podobně jako u skleromyxedému.
U skleredému připadají v úvahu tři možné etiologie:
- Souvislost s monoklonální gamapatií. Typicky jsou postiženi muži středního věku života. Tento typ je předmětem našeho popisu. Ústup této kožní patologie při potlačení tvorby M-Ig opět mluví pro souvislost s monoklonální gamapatií, podobně jako v případech skleromyxedému [24–26].
- Skleredém se může vyskytnout u pacientů s diabetem mellitem a metabolickým syndromem. Tito pacienty mívají obvykle adipositas per magna.
- Skleredém jako následek infekce, obvykle streptokokové. Tento typ obvykle samovolně brzy odezní. Tato reakce je popisována u dětí a mladých žen.
Na našem pracovišti jsme se skleredémem setkali u nemocných se symptomatickým mnohočetným myelomem. Dosažení kompletní remise mnohočetného myelomu vedlo k vymizení kožních projevů skleredému (obr. 4).
![Skleredém indukovaný monoklonálním imunoglobulinem. Po dosažení kompletní remise vymizel a znovu se vyskytl v době recidivy myelomu. Kůže pacienta byla tak tuhá, že nebylo možné vytvořit kožní řasu. Převzato z [24]. Publikace fotografi e se souhlasem pacienta.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/146c75ceb0d6c760ea9ea182d4af2c84.png)
Normolipemická xantomatóza
Xantomatóza je porucha s charakteristickým vznikem žlutých tukových depozit v podkoží. Tato depozita se mohou objevit v různých částech těla, na víčkách, v okolí kloubů a šlach, ve vnitřních orgánech. Jsou tvořena makrofágy obsahujícími velké inkluze lipidů.
Plošné xantomy se mohou objevit u hyperlipidemických pacientů a v těchto případech zřejmě souvisejí s hyperlipidemií. Pokud se ložisko tuk obsahujících makrofágů vyskytuje na víčkách, nazývá se xantelasma (periorbitální plošný xantom), případně pacient může mít i více těchto ložisek na víčkách – vícečetná xantelasmata.
Plošné xantomy se mohou také vyskytnout bez jakékoliv vazby na zvýšené hodnoty tuků v krvi. Ty se pak nazývají diffuse normolipemic plane xanthoma a jsou obvykle asociované s monoklonální gamapatií.
Při tomto typu onemocnění se formují plošné xantomy hlavy, krku, trupu, ramen či končetin. Kožní plaky žlutooranžové barvy (normolipemická xantomatóza) byly popsány u pacientů s myelomem nebo s MGUS, ale také u pacientů s lymfomy, a dokonce i u Castlemanovy choroby. Patofyziologicky zde jde o intracelulární akumulaci imunitních komplexů složených z M-Ig a z lipoproteinů.
Morfologicky je pro normolipemickou xantomatózu charakteristická intracelulární akumulace tukového materiálu v makrofázích, které se hromadí v kůži v okolí vaziva šlach a tvoří ložiska pěnitých buněk neboli foamy cells, přičemž v séru pacienta jsou nezvýšené koncentrace triglyceridů a cholesterolu.
Kožní plošné normolipemické xantomy je třeba odlišit od xantogranulomu, který tvoří granulomy a mívá tmavší zabarvení [27,28].
Na obr. 5 je náš pacient s kožní normolipemickou xantomatózou.

Nekrobiotický xantogranulom
Nekrobiotický xantogranulom je onemocnění projevující se nažloutlými granulomy, které nejčastěji postihují víčka, mohou se ale vyskytnout kdekoliv v těle. Je vázáno na přítomnost M-Ig. Odborná literatura rozlišuje juvenilní xantogranulom, xantogranulom a nekrobiotický xantogranulom. Považujeme za nutné ozřejmit rozdíl mezi těmito klinickými jednotkami.
Pátá WHO klasifikace krevních chorob uvádí v podkapitole Histiocytární a makrofágové neoplazie dominující jednotku juvenilní xantogranulom s typickou formou četných kožních papul a nodulů. Tyto juvenilní xantogranulomy se vyskytují u dětí, v dospělosti se výjimečně objevuje lokalizovaný xantogranulom anebo generalizované onemocnění s xantogranulovými ložisky, které se nazývá Erdheimova-Chesterova choroba. Tyto choroby jsou považovány WHO klasifikací za klonální onemocnění s divergentním biologickým chováním.
Mimoto lékařská literatura popisuje jednotku s názvem nekrobiotický xantogranulom, která není zmíněna v poslední verzi WHO klasifikace krevních chorob.
Nekrobiotický xantogranulom má mírně odlišný histologický obraz od klasického xantogranulomu. Někteří autoři jej považují za reaktivní proces vyvolaný určitým typem M-Ig, protože po potlačení M-Ig regreduje. Jiní autoři ve svých recentních publikacích zařazují nekrobiogický xantogranulom do skupiny vzácných non-Langerhans cell histiocytoses. To je skupinové označení použité ve starší klasifikaci histiocytárních chorob vytvořené odbornou společností Histiocyte Society. Klasifikace Histiocyte Society (na rozdíl od WHO klasifikace) zahrnuje jak reaktivní histiocytózy, tak histiocytózy maligní.
V recentní literatuře je několik větších klinických studií, které se této jednotce věnovaly.
Studie z roku 2020 analyzovala 235 pacientů. U 193 případů (82,1 %) byl přítomný M-Ig. Nejčastěji byl zastoupen M-IgG-k (u 117 pacientů, tedy v 50 % případů). Maligní gamapatie byla přítomna u 59 (25,1 % pacientů). Z této studie vzešla takzvaná Delfská kritéria nekrobiotického xantogranulomu, která jsou uvedena v tab. 4 [29]. V roce 2024 byla další studií tato kritéria verifikována [30].
![Delfská diagnostická kritéria nekrobiotického xantogranulomu zveřejněná v roce 2020 a verifi kovaná v roce 2024 [29,30].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/41a91420dbadd461b1d9c6c79c0ef886.png)
Nekrobiotický xantogranulom v citovaných studiích, a také ve všech popisech případů, vykazuje výraznou asociaci s monoklonální gamapatií a při potlačení této gamapatie má tendenci regredovat. Předpokládanou etiopatogenetickou podstatou je akumulace imunitních komplexů (M-Ig + lipoprotein) v infiltrátech tvořených pěnitými buňkami (foamy cells) s lokální zánětlivou reakcí ve žlutooranžově zabarveném ložisku.
Při histologickém vyšetření jsou nalézány makrofágy, které akumulují lipidy a vytvářejí typická ložiska pěnitých buněk. Dalším morfologickým znakem je nekrobióza (kolagenolýza) v popsaných ložiscích. V ložiscích nekrobiotického xantogranulomu jsou nalézány mimo typické pěnité buňky také epiteloidní buňky, lymfocyty a Toutonovy buňky, odpovídající zánětlivé reakci. Tvorba imunokomplexů může vést k aktivaci komplementu a k poklesu hladiny C4 složky komplementu [29,30].
Termín necrobiosis v případě nekrobiotického xantogranulomu označuje kolagenolýzu. Současná literatura doporučuje tento termín používat pouze v souvislosti s jednotkami, které tento termín mají v názvu: necrobiotic xanthogranuloma a necrobiosis lipoidica, protože zde je to historicky dáno. V jiných případech je doporučeno místo termínu necrobiosis použít více výstižný popis probíhajícího procesu [31].
Nekrobiotický xantogranulom bývá často lokalizován periorbitálně, ale může postihovat i nitro orbity. Pokud je přítomný na víčkách, způsobuje proptózu, blefaroptózu, skleritidu, zvětšení slzných žlázek a omezuje pohyb očí. Nekrobiotický xantogranulom ale může být lokalizován kdekoliv v těle. U jednoho pacienta jsme léčili obtěžující ložiska xantogranulomu v podkoží lýtek, která komprimovala žilní systém, ale i pacienta, který měl ložisko xantogranulomu v játrech, které po dosažení kompletní remise myelomu totálně vymizelo.
Nekrobiotický xantogranulom může ustoupit, pokud se podaří potlačit tvorbu vyvolávajícího M-Ig. Druhá alternativa léčby je podávání IVIG v imunomodulačních dávkách [29–35]. Xantogranulomy asociované s monoklonální gamapatií v oblasti víček zobrazuje obr. 6, xantogranulomy mimo obličej pak obr. 7.

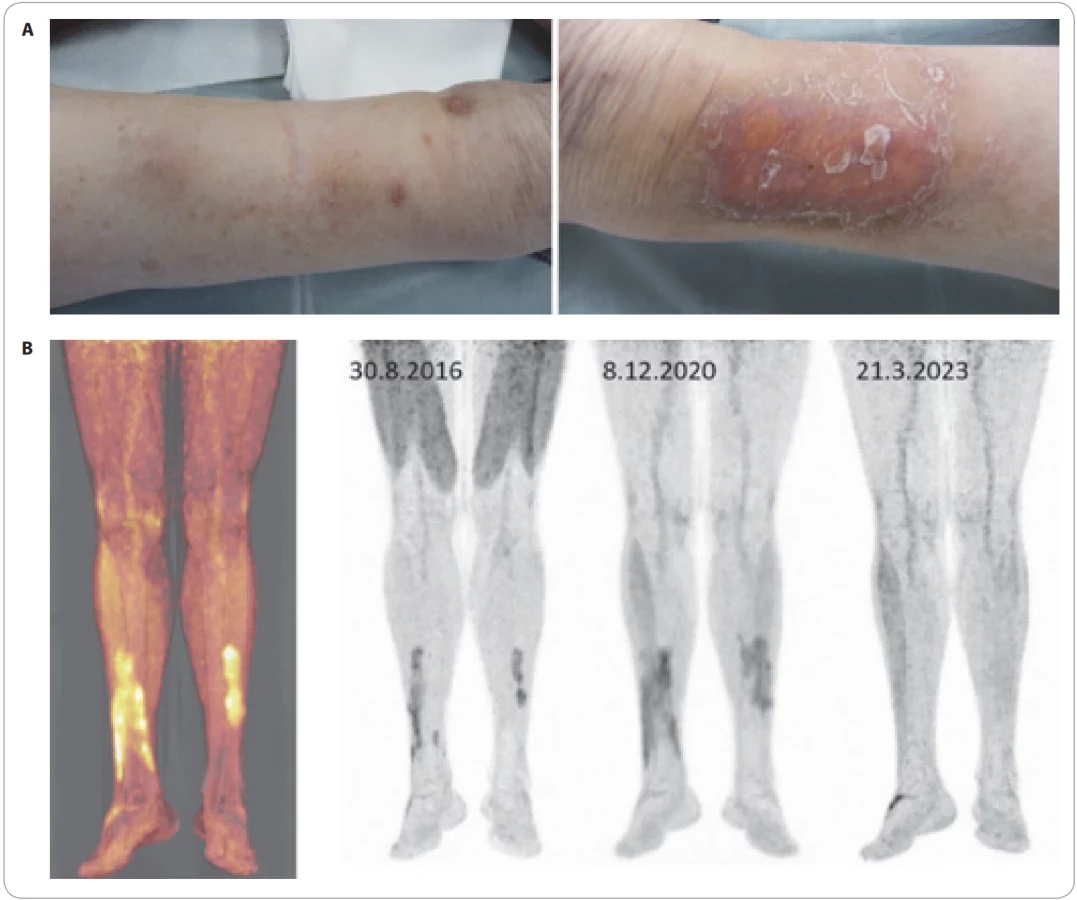
Syndrom Schnitzlerové
Syndrom Schnitzlerové je vzácné multisystémové onemocnění středního věku, které způsobuje chronické či opakované výsevy kopřivky, periodické subfebrilie či febrilie, bolesti kostí a někdy i kloubů a patologickou únavu. Onemocnění je spjato s dysregulací cytokinů (nadprodukce interleukinu-1), a proto je řazeno také mezi autoinflamatorní choroby. Obligátní přítomnost M-Ig jej ale také začleňuje mezi monoklonální gamapatie. Syndrom Schnitzlerové je asociován ve většině případů s M-Ig typu IgM, výjimečně byl asociován s jiným typem M-Ig. Na etiopatogenezi se podílí autoinflamatorní mechanizmy s nadprodukcí cytokinů. Pokud léčba monoklonální gamapatie dosáhne parciální remise, tak se zmenší dávky anakinry (antagonisty receptoru interleukinu-1) nutné k potlačení projevů nemoci. To dokazuje souvislost s monoklonální gamapatií, více informací o etiopatogenezi této nemoci však nemáme.
Kožním projevem syndromu Schnitzlerové jsou ataky kopřivky. Její výsev je spojen s dalšími projevy, které tvoří kritéria této jednotky, jak uvádí tab. 5 [36,37].
![Kritéria syndromu Schnitzlerové [27,28,36,37].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/f716119b69533a0fad0eb7d40f3d7ee9.png)
Na možnost syndromu Schnitzlerové je vhodné vzpomenout si u každého člověka s chronicky recidivující urtikarií a s projevy systémové zánětlivé reakce. Pacienti s tímto syndromem jsou léčeni obvykle hematology, protože monoklonální gamapatie typu IgM se u nich často transformuje ve Waldenströmovu makroglobulinemii, takže je na ošetřujícím hematologovi, aby tento vývoj včas rozpoznal a včas začal s cílenou léčbou tohoto onemocnění [36–40].
Léčbou volby je preparát anakinra nebo jiné léky blokující interleukin-1. Léčba potlačující monoklonální gamapatie se používá až při transformaci MGCS do maligní choroby, obvykle při transformaci nemaligní IgM gamapatie do Waldenströmovy makroglobulinemie. Kožní projevy u pacientů se syndromem Schnitzlerové ilustruje obr. 8.

TEMPI syndrom
TEMPI syndrom je vzácné multisystémové onemocnění s kožními projevy. Název nemoci je akronym vytvořený z typických symptomů této nemoci: teleangiektazie, erytrocytóza se zvýšenou hladinou erytropoetinu, monoklonální gamapatie, perinefritické kolekce tekutiny a intrapulmonální arteriovenózní spojky (shunty). Obligátní přítomnost M-Ig jej řadí do skupiny MGCS.
TEMPI syndrom je nejčastěji asociován s monoklonální gamapatií typu IgG-k, jsou ale popsány případy asociované s typem IgA a na našem pracovišti jsme popsali pacienta s TEMPI syndromem při Waldenströmově makroglobulinemii. Pacienti s touto diagnózou obvykle dlouze unikají rozpoznání diagnózy a jsou vedeni jako sekundární polyglobulie. Pro časnou diagnostiku je nutné u každé erytrocytózy s normální či zvýšenou hodnotou erytropoetinu vyšetřit M-Ig a při jeho prokázání zaměřit diferenciální diagnostiku na tuto chorobu.
Stejně tak nově se objevující teleangiektazie na kůži pacienta by měly vést k vyšetření M-Ig. Při průkazu monoklonální gamapatie u osoby s polyglobulií anebo s nově vznikajícími teleangiektaziemi je nutno diferenciálně diagnostiky vyšetřit možnost POEMS anebo TEMPI syndromu. Obě tyto jednotky způsobují polyglobulii, ale také kožní projevy typu teleangiektazií a hemangiomů.
Typické projevy ilustrují obrazy našeho prvního pacienta s touto chorobou (obr. 9) a podrobnosti o této nemoci uvádí citovaná literatura. Pro diagnostiku byla přijata kritéria viz tab. 6. Dle literatury vede eliminace M-Ig i k eliminaci těchto projevů [41,42].

![Kritéria TEMPI syndromu, které v roce 2020 zveřejnil David Sykes [41].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/eab2cfe67f5b3e448e0a909d0a253fb2.png)
AESOP syndrom
AESOP syndrom je vzácné onemocnění charakteristické lividními či erytematózními kožními plochami nad ložiskem kostního plazmocytomu. Název nemoci je opět akronym vytvořený z počátečních písmen typických symptomů nemoci: adenopathy and extensive skin patch overlying a plasmacytoma. Přeloženo: adenopatie (obvykle regionální) s větší kožní erytémovou morfou nad ložiskem kostního plazmocytomu. AESOP syndrom je velmi vzácnou klinickou jednotkou, která byla akceptována do 5. vydání WHO klasifikace maligních krevních chorob. AESOP je dobře rozpoznatelným kožním syndromem, je charakterizován pomalu expandujícím kožním ložiskem, pod nímž se v hloubi ukrývá solitární kostní plazmocytom.
Předpokládá se, že kožní změny souvisejí s lokální nadprodukcí VEGF solitárním plazmocytomem.
Patologické kožní ložisko má rudou barvu, je hladké a pod kůží prosvítají zmnožené cévy. To je považováno za klasickou variantu. Alternativou mohou být indurované červenohnědé plaky vypadající jako pomerančová kůže. Tato podoba AESOP syndromu se označuje jako morphea-like variant. Typickými lokalizacemi je kůže nad sternem, klavikulou, na bocích a v oblasti hypochondria. Většina pacientů má také zvětšené regionální uzliny. Histopatologické vyšetření kožního ložiska prokáže proliferaci kapilár, smíšenou dermální infiltraci a mucinová depozita. Asi 40 % pacientů s EASOP syndromem při pozdějším přešetřování splní kritéria POEMS syndromu, pokud nejsou včas a intenzivně léčeni [43–45]. Kritéria AESOP syndromu uvádí tab. 7 a kožní projevy ilustruje obr. 10.

![Pomalu se zvětšující hnědavý zvlněný plak barvy pomerančové slupky (peau d’orange) lokalizovaný nad plazmocytomem v žebru [45]. Nemoc diagnostikoval a léčil prof. Dr. Dan Lipsker ve Štrasburku. Autor popsal více podobných případů v odborné literatuře a pro časopis Klinická onkologie poskytl zatím nepublikovanou fotografi i kožního erytému u pacienta se AESOP syndromem.](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/90a3c9b55792db2c72a737be86a9fc8e.png)
Pokud je stanovena diagnóza AESOP syndromu, je nutné další šetření jako u každého solitárního plazmocytomu. V případě potvrzení solitárního plazmocytomu a vyloučení mnohočetného myelomu je základem léčby kurativní radioterapie cílená na ložisko plazmocytomu.
AESOP syndrom je často provázen závažnou neuropatií, první popis se datuje z roku 1938 a další popisy se začaly objevovat zpočátku v neurologické literatuře, protože přidruženým problémem byla neuropatie.
Kryoglobulinemie
Kryoglobulinemie je termín pro přítomnost bílkovin, které kryoprecipitují v cévním řečišti při poklesu teploty pod fyziologické rozmezí. Kryoglobulinemie se klasifikuje dle komponenty, která precipituje. Klinické příznaky závisí na teplotě, při níž kryoprotein gelifikuje.
Kryoglobulin I. typu je tvořen M-IgM nebo jiným typem M-Ig, který samostatně precipituje při ochlazení, aniž by se specificky vázal na jiné bílkoviny. Porucha prokrvení tkání na podkladě kryoglobulinu může vést i k trvalému poškození, nejtěžšími projevy jsou kožní ulcerace a nekrózy a případně poškození ledvin.
Kryoglobulin II. typu je definován jako M-Ig, který se v chladu váže na Fc-fragmenty polyklonálních imunoglobulinů jiných tříd. Typicky je to M-IgM, který se váže na Fc-fragmenty polyklonálních imunoglobulinů typu IgG. M-IgM má v tomto případě charakter revmatoidního faktoru. Tato nemoc je nazývána smíšená kryoglobulinemie (mixed cryoglobulinaemia). Kryoglobulinemie II. typu je imunokomplexová choroba, a proto má podstatně pestřejší projevy než kryoglobulinemie I. typu. Ke kožním projevům kryoglobulinemie II. typu patří livedo retikularis. Pacienti mohou mít hyperpigmentace na dolních končetinách jako následky opakované purpury. Tyto hyperpigmentace mohou tvořit i hmatná kožní zduření. Taktéž ulcerující ložiska na kůži patří k možným projevům. Všechny tyto projevy jsou důsledkem imunokomplexové vaskulitidy [46–53].
Kryoglobulin III. typu (synonymem je polyklonální kryoglobulin) je tvořen pouze polyklonálními imunoglobuliny s tepelnou charakteristikou kryoglobulinů. Kryoglobulinemie III. typu je pozorována nejčastěji v souvislosti s hepatitidou C, která vyvolává lymfoproliferativní odpověď. Hepatitida C se může transformovat do maligní lymfoproliferace, frekvence přechodu dosahuje až 10 %. Typické projevy této formy kryoglobulinemie jsou artralgie a kožní purpura, podobně jako u II. typu kryoglobulinemie. Kryoglobulinemie II. typu stejně jako kryoglobulinemie III. typu vytváří kryoprecipitáty, které jsou směsí imunoglobulinů IgG a IgM. Proto se oběma typům říká také smíšená kryoglobulinemie. Podrobnosti o analýze kryoglobulinu uvádí Kušnierová [46]. Projevy kryoglobulinemie shrnuje tab. 8. Pestrost projevů smíšené kryoglobulinemie zdůrazňuje schéma 2.

(hematoonkologičtí pacienti mají převážně typ I, revmatologičtí pacienti a pacienti z infekčních ambulancí/klinik převážně typy II
a III).
** Pokud je termín „oligoklonální“ použit ve smyslu „dva a více různých monoklonálních imunoglobulinů“, vyčleňuje se někdy kryoglobulin takového složení do podkategorie IIB.
CMV – cytomegalovirus, EBV – virus Epsteina-Barrové, HBV – virus hepatitidy B, HCV – virus hepatitidy C, HIV – virus získané lidské
imunodefi cience, MGUS – monoklonální gamapatie nejistého významu, NHL – non-hodgkinský lymfom, SLE – systémový lupus erythematosus, RA – revmatoidní artritida

Několik projevů kryoglobulinemie u našich pacientů ilustruje obr. 11. Akutní léčbou, zvláště u kryoglobulinemie typu IgM, je plazmaferéza. Hlavní kauzální léčbou je pak potlačení buněčné populace, která kryoglobulin produkuje [54].

Cutis laxa
Termínem cutis laxa se označují vrozená či získaná onemocnění, která se projevují lokální či generalizovanou ztrátou elasticity kůže, takže ta začne být povislá a vytváří řasy. Nemoc způsobuje lokální či generalizovanou degradaci elastických vláken. Upřednostníme latinský termín, český ekvivalent „volná kůže“ jsme v literární databázi medvik.cz nenašli.
V případech získané formy choroby zvané cutis laxa tvoří monoklonální gamapatie 27 %. Cutis laxa je popsána jak v souvislosti s mnohočetným myelomem, tak v souvislosti s dalšími monoklonálními gamapatiemi, včetně nemoci zvané choroba z ukládání lehkých a těžkých řetězců (heavy and light-chain deposition diseases). Zahraniční literatura popisuje jednotlivé případy i malé série pacientů se získanou formou cutis laxa. V domácí literatuře není zatím popis případu cutis laxa, který by souvisel s monoklonální gamapatií, zatímco hereditární případy jsou popsány v domácí pediatrické literatuře [55–58].
Kožní změny s nižší pravděpodobností souvislosti s M-Ig
Ze skupiny kožních změn s nižší asociací s monoklonální gamapatií uvedeme pouze zmínku o IgA pemphigu.
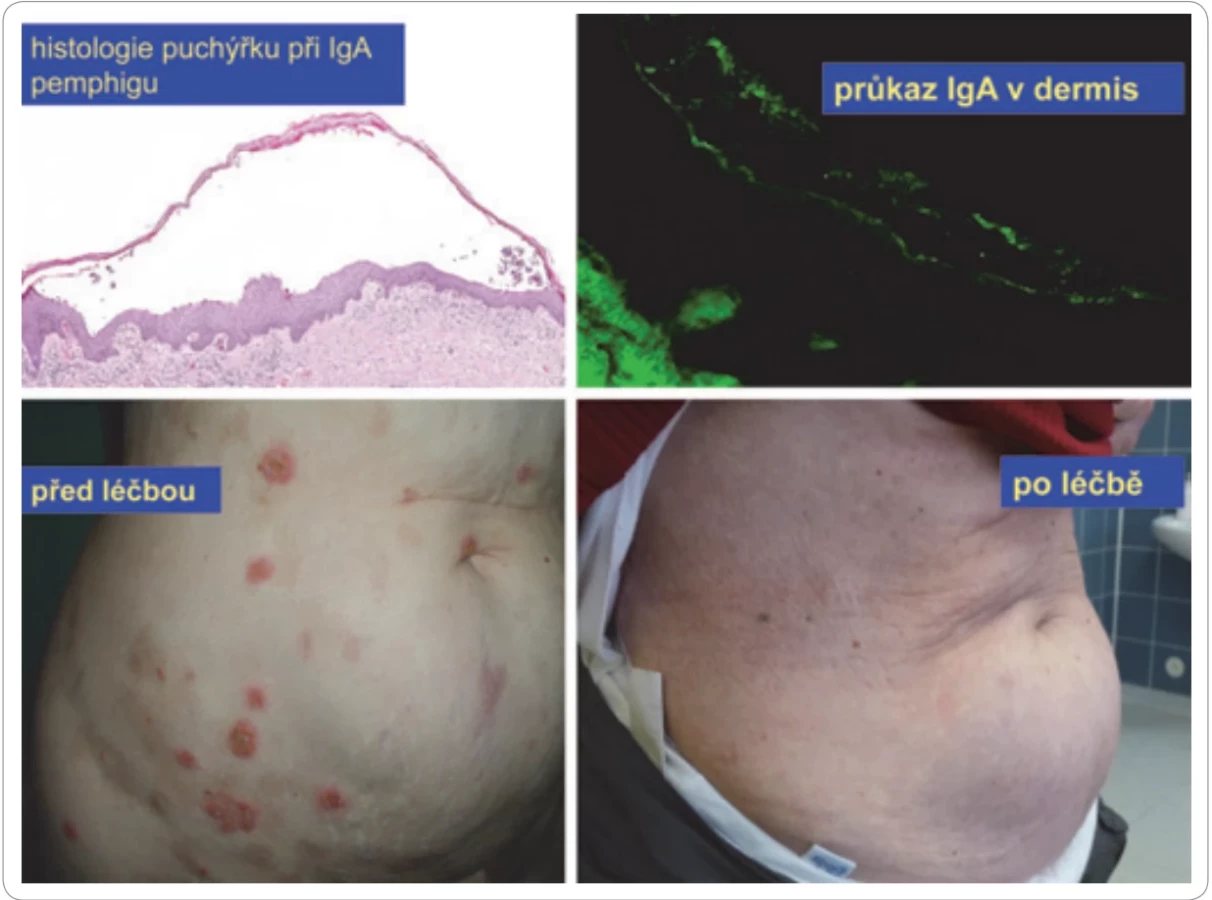
Subkorneální pustulózní dermatóza neboli IgA-pemphigus
Pemphigus je skupina vzácných autoimunitních puchýřnatých chorob, u kterých dochází k tvorbě puchýřů (buly) jak na kůži, tak i na sliznicích. Jméno nemoci je odvozeno z řeckého slova pemphix, což znamená puchýř. Puchýře vznikají v epidermis poškozením intraepidermální adheze, spojů mezi buňkami (akantolýza). Autoprotilátky způsobující toto onemocnění jsou namířeny proti strukturám mezibuněčných spojů v epidermis. Ve vzácných případech může být touto protilátkou poškozující kůži také M-Ig, tvořený benigní či maligní plazmocelulární populací.
Setkali jsme se s pacientkou, které trpěla pemphigem po mnoho let, zatímco byla v naší ambulanci sledována pro monoklonální gamapatii nejistého významu (MGUS) typu IgA. MGUS se u této pacientky postupně vyvíjel a transformoval do symptomatického mnohočetného myelomu. Léčba myelomu u této pacientky dosáhla kompletní remise (úplné vymizení M-Ig, potvrzené negativní imunofixační elektroforézou) a překvapivě vymizely morfy pemphigu. Po letech došlo k recidivě myelomu, a ta byla opět spojena s recidivou pemphigu. Recidiva pemphigu související s recidivou myelomu jasně potvrzuje souvislost.
Ve světové medicínské literatuře jsou popsány četné případy, kdy touto autoprotilátkou byl M-IgA. Léčba, která úspěšně potlačila tvorbu M-IgA, zastavila také tvorbu nových puchýřků a pustul. Pro stanovení diagnózy je přínosný morfologický průkaz imunoglobulinových IgA depozit intercelulárně v epidermis s průkazem toho samého typu imunoglobulinu (příp. toho samého izotypu M-Ig a typu lehkého řetězce, jako má cirkulující M-Ig). Pemphigus je autoimunitní onemocnění kůže způsobené autoprotilátkami a v některých případech mohou kůži poškozovat právě molekuly M-Ig, obvykle typu IgA [59–62]. Bulózní kožní poškození, tzv. IgA pemphigus, zobrazuje obr. 12.
Závěr
Kožní změny mají velmi často vazbu na různá systémová onemocnění. V textu jsou popsány jednotky etiopatogeneticky úzce svázané s monoklonální gamapatií [63]. Je důležité, aby dermatologové tyto jednotky znali a vždy u nich požádali o vyšetření M-Ig a FLC. Pokud prokáží přítomnost M-Ig a/nebo patologické koncentrace či poměr FLC, pak je důležité, aby stav těchtp pacientů probrali s hematology, kteří jim mohou poskytnout cílenou léčbu po schválení plátcem zdravotní péče.
Dedikace
Publikace byla vytvořena na podporu těchto aktivit: MZ ČR – RVO (FNBr, 65269705) a institucionální aktivity MOÚ: MZ ČR – RVO (MOÚ, 00209805).
Sources
1. Cetkovská P, Pizinger K, Štork J. Kožní změny u interních onemocnění. Praha: Grada 2010.
2. Lipsker D. Monoclonal gammopathy of cutaneous significance: review of a relevant concept. J Eur Acad Dermatol Venereol 2017; 31 (1): 45–52. doi: 10.1111/jdv.13847.
3. Ríos-Tamayo R, Paiva B, Lahuerta JJ et al. Monoclonal gammopathies of clinical significance: a critical appraisal. Cancers (Basel) 2022; 14 (21): 5247. doi: 10.3390/cancers14215247.
4. Claveau JS, Wetter DA, Kumar S. Cutaneous manifestations of monoclonal gammopathy. Blood Cancer J 2022; 12 (4): 58. doi: 10.1038/s41408-022-00661-1.
5. Berk-Krauss J, Rosenbach M. Monoclonal gammopathy of cutaneous significance. J Am Acad Dermatol 2025; 93 (4): 1042–1048. doi: 10.1016/j.jaad.2025.05.1432.
6. Iberri D, Liedtke M. MGCS: where do we stand today? Hematology Am Soc Hematol Educ Program 2024; 2024 (1): 482–488. doi: 10.1182/hematology.2024000572.
7. Flodrová P, Pika T, Flodor P. AL amyloidóza v obrazech. Trans Hematol Dnes 2014; 20 (3): 76–80.
8. Pika T. Diagnostika a léčba systémové AL amyloidózy: doporučení vypracovaná Českou myelomovou skupinou (CMG), Myelomovou sekcí České hematologické společnosti ČLS JEP. Trans Hematol Dnes 2022; 28 (Suppl 1): 6–40.
9. Khogeer A, Dupuy A. Cutaneous macroglobulinosis with Waldenström macroglobulinemia and Bing-Neel syndrome: a case report. JAAD Case Rep 2025; 61 : 82–84. doi: 10.1016/j.jdcr.2025.04.009.
10. Dispenzieri A. POEMS syndrome: update on diagnosis, risk-stratification, and management. Am J Hematol 2023; 98 (12): 1934–1950. doi: 10.1002/ajh.27081.
11. Lee K, Kourelis T, Tschautscher M et al. Capillary leak phenotype as a major cause of death in patients with POEMS syndrome. Leukemia 2025; 39 (3): 703–709. doi: 10.1038/s41375-024-02489-z.
12. Gallardo-Pérez MM, Negrete-Rodríguez P, Gertz MA et al. The Latin-American experience in POEMS syndrome: a study of the GELAMM (Grupo de Estudio Latinoamericano de Mieloma Múltiple). Acta Haematol 2025; 148 (3): 249–257. doi: 10.1159/000540890.
13. Talbot A, Forgeard N, Vaugeois T et al Teclistamab for heavily pretreated relapsed / refractory POEMS syndrome. Haematologica 2025. doi: 10.3324/haematol.2025.287606.
14. Krausová A, Ježková J. Diabetologie, metabolismus, endokrinologie, výživa. 2023 [online]. Dostupné z: https: //www.tigis.cz/casopisy/pro-lekare/psychiatrie11/item/1485-dmev-2-2023.
15. Minařík J, Ščudla V, Bačovský J et al. POEMS syndrom. Onkologie 2011; 5 (3): 151–154.
16. Ueda F, Okuda M, Aburano H et al. Cranial pachymeningeal involvement in POEMS syndrome: evaluation by pre - and post-contrast FLAIR and T weighted imaging. Magn Reson Med Sci 2017; 16 (3): 231–237. doi: 10.2463/mrms.mp.2015-0014.
17. Briani C, Manara R, Lessi F et al. Pachymeningeal involvement in POEMS syndrome: dramatic cerebral MRI improvement after lenalidomide therapy. Am J Hematol 2012; 87 (5): 539–541. doi: 10.1002/ajh.23148.
18. Ziff OJ, Hoskote C, Keddie S et al. Frequent central nervous system, pachymeningeal and plexus MRI changes in POEMS syndrome. J Neurol 2019; 266 (5): 1067–1072. doi: 10.1007/s00415-019-09233-z.
19. Adam Z, Řehák Z, Keřkovský M et al. Monoclonal gammopathy of clinical significance with osteosclerotic lesions – a case report and a literature review. Klin Onkol 2024; 37 (3): 209–219. doi: 10.48095/ccko 2024209.
20. Li ZY, Chen JJ, Lu FY et al. Non-POEMS osteosclerotic multiple myeloma: clinical characteristics and differential diagnosis. J Bone Oncol 2024; 45 : 100595. doi: 10.1016/j.jbo.2024.100595.
21. Rongioletti F, Merlo G, Cinotti E et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol 2013; 69 (1): 66–72. doi: 10.1016/j.jaad.2013.01.007.
22. Theves F, Lahuna C, Mahévas T et al. Plasma cell-directed therapies induce profound clinical and durable responses in patients with severe or relapsed/refractory scleromyxedema. J Eur Acad Dermatol Venereol 2025; 39 (5): 1011–1016. doi: 10.1111/jdv.20257.
23. Di Battista M, Grosso M, Marciano A et al. Efficacy of intravenous immunoglobulins in severe scleromyxedema dysphagia assessed by oesophageal scintigraphy. Clin Exp Rheumatol 2024; 42 (8): 1706. doi: 10.55563/clinexprheumatol/2a5goh.
24. Adam Z, Szturz P, Krejčí M et al. Monoclonal immunoglobulin (M-Ig) and skin diseases from the group of mucinoses – scleredema adultorum Buschke and scleromyxedema. Description of four cases and an overview of therapies. Vnitr Lek 2015; 61 (12): 1072–1087.
25. Knobler R, Geroldinger-Simić M, Kreuter A et al. Consensus statement on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxoedema and scleroedema. J Eur Acad Dermatol Venereol 2024; 38 (7): 1281–1299. doi: 10.1111/jdv.19937.
26. Král Z, Krejčí M, Pour L et al. Vzácné kožní změny asociované s monoklonální gamapatií: skleredém, skleromyxedém a IgA pemfigus – popis pěti případů a přehled léčebných možností. Trans Hematol Dnes 2020; 26 (3): 226–235.
27. Cohen YK, Elpern DJ. Diffuse normolipemic plane xanthoma associated with monoclonal gammopathy. Dermatol Pract Concept 2015; 5 (4): 65–67. doi: 10.5826/dpc.0504a16.
28. Adam Z, Szeligová L, Krejčí M et al. Diffuse plane normolipemic xanthomatosis and necrobiotic xanthogranuloma associated with monoclonal gammopathy – determining the disease stage with PET-CT and treatment experience. Two case studies and literature review. Vnitr Lek 2010; 56 (11): 1158–1168.
29. Nelson CA, Zhong CS, Hashemi DA et al. A multicenter cross-sectional study and systematic review of necrobiotic xanthogranuloma with proposed diagnostic criteria. JAMA Dermatol 2020; 156 (3): 270–279. doi: 10.1001/jamadermatol.2019.4221.
30. Bassir F, Valido K, Maciejewski KR et al. Validation of the delphi consensus diagnostic criteria for necrobiotic xanthogranuloma. JAMA Dermatol 2024; 160 (12): 1361–1362. doi: 10.1001/jamadermatol.2024.3198.
31. Gupta S, Joshi R, Chikhalkar S et al. Understanding the term „necrobiosis“. Indian Dermatol Online J 2024; 15 (3): 552–555. doi: 10.4103/idoj.idoj_674_23.
32. Adam Z, Veselý K, Motyčková I et al. Eylids with yellow granulomas and cough – periocular xanthogranuloma associated with adult-onset asthma. A case study and an overview of clinical forms of juvenile xanthogranuloma and its therapy. Vnitr Lek 2012; 58 (5): 365–377.
33. Adam Z, Řehák Z, Adamová Z et al. Slow increase of bilirubin concentration during administration of lenalidomide, bortezomid and dexamethasone for multiple myeloma (unmasking previously undiagnosed Gilbert syndrome) and disappearance of necrobiotic xanthogranuloma after complete remission of multiple myeloma. Klin Onkol 2022; 35 (4): 315–322. doi: 10.48095/ccko2022315.
34. Adam Z, Pour L, Řehák Z et al. Complete remission of necrobiotic xanthogranuloma after disappearance of monoclonal immunoglobulin induced by bortezomib, lenalidomid and dexamethasone. Vnitr Lek 2021; 67 (6): 352–356.
35. Steinhelfer L, Kühnel T, Jägle H et al. Systemic therapy of necrobiotic xanthogranuloma: a systematic review. Orphanet J Rare Dis 2022; 17 (1): 132. doi: 10.1186/s13023-022-02291-z.
36. Simon A, Asli B, Braun-Falco M et al. Schnitzler‘s syndrome: diagnosis, treatment, and follow-up. Allergy 2013; 68 (5): 562–568. doi: 10.1111/all.12129.
37. Lipsker D, Veran Y, Grunenberger F et al The Schnitzler syndrome. Four new cases and review of the literature. Medicine (Baltimore) 2001; 80 (1): 37–44. doi: 10.1097/00005792-200101000-00004.
38. Adam Z, Šedivá A, Koukalová R et al. Schnitzlers syndrome differential diagnostics, and overview of therapeutic options and description of 5 cases treated with anakinra. Vnitr Lek 2016; 62 (9): 713–727.
39. Adam Z, Tomíška M, Řehák Z et al. Transformation of IgM-MGUS into Waldenström’s macroblobulinemia in two of six patients treated for Schnitzler’s syndrome. Vnitr Lek 2021; 67 (E–3): 15–23.
40. Adam Z, Mayer J, Pour L et al. Autoinflamatorní choroby se symptomy, které připomínají maligní krevní choroby – syndrom Schnitzlerové, Stillova choroba dospělých, SAPHO a VEXAS syndrom. Trans Hematol Dnes 2024; 30 (3): 151–168. doi: 10.48095/cctahd2024prolekare.cz13.
41. Sykes DB, O‘Connell C, Schroyens W. The TEMPI syndrome. Blood 2020; 135 (15): 1199–1203. doi: 10.1182/blood.2019004216.
42. Adam Z, Leová L, Patočková M et al. Description of TEMPI syndrome in Waldenström’s macroglobulinemia. Klin Onkol 2025; 38 (4): 283–301. doi: 10.48095/ccko 2025283.
43. Shaker N, Sangueza OP. Concomitant AESOP and POEMS syndrome in the context of osseous and cutaneous plasmacytomas: answer. Am J Dermatopathol 2023; 45 (12): 852–853. doi: 10.1097/DAD.0000000000 002561.
44. Yan W, Lv R, Xu J et al. Adenopathy and extensive skin patch overlying a plasmacytoma (AESOP) syndrome: a case report and literature review. Ann Hematol 2024; 103 (1): 339–341. doi: 10.1007/s00277-023-05465-2.
45. Lipsker D, Rondeau M, Massard G et al. The AESOP (adenopathy and extensive skin patch overlying a plasmacytoma) syndrome: report of 4 cases of a new syndrome revealing POEMS (polyneuropathy, organomegaly, endocrinopathy, onoclonal protein, and skin changes) syndrome at a curable stage. Medicine (Baltimore) 2003; 82 (1): 51–59. doi: 10.1097/00005792-200301000-00005.
46. Kušnierová P. Laboratorní diagnostika monoklonálních gamapatií. Ostravská univerzita 2022.
47. Duggal S, Kattamuri LVP, Toutoungy M et al. Exploring cryoglobulinemia‘s clinical Odyssey: a case series. EJHaem 2025; 6 (2): e70029. doi: 10.1002/jha2.70029.
48. Codes-Méndez H, Jeria S, Park HS et al. Clinical and serological profiles in cryoglobulinemia: analysis of isotypes and etiologies. J Clin Med 2024; 13 (20): 6069. doi: 10.3390/jcm13206069.
49. Sečníková Z. Kryoglobulinémie a kryofibrinogémie. Cesk Dermatovenerol 2018; 8 (3): 184–188.
50. Čermáková Z, Gottwaldová J. Kryoglobulinémie a její rizika při laboratorním vyšetřování – kazuistika. Klin Biochem Metab 2009; 17 (2): 79–80.
51. Adam Z, Zdražilová Dubská L, Pour L et al. Kryoglobulinémie z úhlu pohledu jednotlivých medicínských odborností. Trans Hematol Dnes 2024; 30 (4): 213–223. doi: 10.48095/cctahd2024prolekare.cz19.
52. Jančová E, Tesař V, Stejskalová A et al. Postižení ledvin u kryoglobulinémie. Prakt Lek 1998; 78 (3): 109–114.
53. Minařík J, Pika T, Bačovská J et al. Kryoglobulinemická vaskulitida u nemocného s mnohočetným myelomem. Intern Med Praxi 2012; 14 (12): 478–480.
54. Battaglini J, Henderson J, Sanchorawala V et al. Teclistamab therapy for refractory type 1 cryoglobulinemia. Haematologica 2025; 110 (8): 1891–1893. doi: 10.3324/haematol.2025.287313.
55. Khodeir J, Ohanian P, Feghali J. Acquired cutis laxa: a clinical review. Int J Dermatol 2024; 63 (10): 1334–1356. doi: 10.1111/ijd.17338.
56. Nota NM, Westerman M. Severe cutis laxa caused by immunoglobulin M gammopathy. Br J Haematol 2022; 196 (4): 802. doi: 10.1111/bjh.17912.
57. Gillion V, Vekemans MC, Rinsant A et al. Acquired cutis laxa from heavy chain deposition disease. Kidney Int 2022; 102 (6): 1432–1433. doi: 10.1016/j.kint.2022. 08.035.
58. Jachiet M, Harel S, Saussine A et al. Cutis laxa associated with monoclonal gammopathy: 14 new cases and review of the literature. J Am Acad Dermatol 2018; 79 (5): 945–947. doi: 10.1016/j.jaad.2018.03.039.
59. Simionescu O, Tudorache SI. Autoimmune pemphigus: difficulties in diagnosis and the molecular mechanisms underlying the disease. Front Immunol 2025; 16 : 1481093. doi: 10.3389/fimmu.2025.1481093.
60. Adam Z, Feit J, Krejčí M et al. IgA pemphigus accompanying multiple myeloma has disappeared following the treatment with bortezomib (Velcade), cyclophosphamide and dexamethasone. Case study and literature review. Vnitr Lek 2009; 55 (10): 981–990.
61. Koga H, Tsutsumi M, Teye K et al. Subcorneal pustular dermatosis-type IgA pemphigus associated with multiple myeloma: a case report and literature review. J Dermatol 2023; 50 (2): 234–238. doi: 10.1111/1346-81 38.16516.
62. Kridin K, Patel PM, Jones VA et al. IgA pemphigus: a systematic review. J Am Acad Dermatol 2020; 82 (6): 1386–1392. doi: 10.1016/j.jaad.2019.11.059.
63. Dispenzieri A. Monoclonal gammopathies of clinical significance. Hematology Am Soc Hematol Educ Program 2020; 2020 (1): 380–388.
64. Mahévas T, Arnulf B, Bouaziz J-D. et al Plasma cell-directed therapies in monoclonal gammopathy-associated scleromyxedema. Blood 2020; 135 (14): 1101–1110. doi: 10.1182/blood.2019002300.
Labels
Paediatric clinical oncology Surgery Clinical oncologyArticle was published in
Clinical Oncology

2026 Issue 1
Most read in this issue
- Rozsah reportování při vyšetření predispozice k nejčastějším solidním nádorům dospělého věku pomocí sekvenování nové generace
- Výsledky léčby Hodgkinova lymfomu v České republice
- Kožní nemoci způsobené monoklonálním imunoglobulinem a/nebo volnými lehkými řetězci (monoklonální gamapatie kožního významu)
- Logopedická intervence u pacientky s mnohočetným myelomem a dysfagií – případová studie z klinické praxe