
Hyperlipoproteinemie a dyslipidemie jako vzácná onemocnění: diagnostika a léčba
Authors:
Richard Češka 1; Tomáš Štulc 1; Lucie Votavová 2; Lucie Schwarzová 1; Martina Vaclová 1; Tomáš Freiberger 3
Authors‘ workplace:
III. interní klinika 1. LF UK a VFN v Praze
1; ScreenPro FH, z. s., Praha
2; Centrum kardiovaskulární a transplantační chirurgie, Brno
3
Published in:
Vnitř Lék 2016; 62(11): 887-894
Category:
Reviews
Overview
Samozřejmě, že hyperlipoproteinemie (HLP) a dyslipidemie (DLP) vnímáme především jako onemocnění masového výskytu, jsou dávána do souvislosti s pandemií kardiovaskulárních onemocnění (KVO) jako nejvýznamnější rizikové faktory (RF). Je to jistě pravda a HLP či DLP celkem postihují desítky procent dospělé populace. Nelze ale přehlížet fakt, že existují (převážně vrozené, geneticky podmíněné) poruchy tukového metabolizmu, které sice formálně nemají statut vzácných onemocnění, nicméně podmínky pro zařazení mezi vzácná onemocnění bohatě splňují. Naše sdělení se jen přehledově dotkne vzácných HLP podle dominující poruchy tukového metabolizmu, tedy uvedeme vzácné primární hypercholesterolemie, primární hypertriglyceridemie a vzácné primární smíšené HLP. U těchto onemocnění došlo v posledních letech k neuvěřitelnému pokroku především v oblasti přesné identifikace genetického defektu a mechanizmu vzniku poruchy, nicméně každé z těchto onemocnění by si vyžádalo prostor pro samostatný článek, bohužel ale mimo oblast klinické interny. Podrobněji se tedy budeme zabývat homozygotní familiární hypercholesterolemií (FH), částečně pak i „těžkou“ heterozygotní FH a v další části pak deficitem lipoproteinové lipázy, tedy nemocemi, které jednak představují pro své nositele extrémní, až fatální riziko v mladém věku, jednak je k jejich léčbě možno využít nových možností. Internista by pak měl vědět, kdy na tato onemocnění pomýšlet, jaké jsou hlavní příznaky, kam nemocného odeslat a jak ho léčit. Krátce bude zmíněna i dysbetalipoproteinemie (HLP typ III). Homozygotní FH se vyskytuje ve frekvenci 1 : 1 000 000 (možná i častěji, 1 : 160 000), je charakterizovaná těžkou izolovanou hypercholesterolemií (celkový cholesterol obvykle 15 i více mmol/l), xantomatózou a především velmi časnou manifestací KVO. Výjimkou nejsou infarkty myokardu i v dětském věku. V léčbě představují základ vysoko dávkované statiny, statiny v kombinaci s ezetimibem a nyní nově i PCSK9 inhibitory. Velkým příslibem pro nemocné je pak lomitapid a částečně i mipomersen. Agresivní léčbu představuje potom LDL-aferéza. Deficit lipoproteinové lipázy (HLP typ I) je charakterizován především těžkou hypertriglyceridemií, mléčným zbarvením séra, xantomatózou. Fatální komplikací je potom akutní recidivující pankreatitida. V léčbě je zásadní dieta, nicméně ani ta nestačí ke zvládnutí geneticky podmíněné poruchy. Jedinou schválenou léčbou je genová terapie. Experimentálně jako terapie „off label“ je kazuisticky používán s efektem lomitapid. Sami máme v této experimentální terapii zkušenost. Dysbetalipoproteinemie je vrozenou poruchou metabolizmu lipoproteinů, charakterizovanou vysokou hladinou cholesterolu (C) i triglyceridů (TG). Podkladem tohoto onemocnění je defekt genu pro apolipoprotein E. Klinicky se manifestuje xantomatózou, především ale předčasnou manifestací aterosklerózy (spíše periferní než koronární).
Klíčová slova:
deficit lipoproteinové lipázy – dysbetalipoproteinemie – familiární hypercholesterolemie – genová terapie – homozygotní FH – LDL-aferéza – lomitapid – mipomersen – PCSK9 inhibitory – rare diseases/vzácná onemocnění
Vzácná onemocnění
Oficiálně jsou vzácná onemocnění (rare diseases) definována jako život ohrožující nebo závažná chronická onemocnění, jejichž prevalence je menší než 5 osob z 10 000 a která jsou spojená s významným nárůstem morbidity a mortality nebo značnou redukcí kvality života postižených (definice Evropské komise pro veřejné zdraví). V současnosti je známo kolem 8 000 druhů vzácných onemocnění postihujících jenom v rámci Evropské unie přibližně 6–8 % populace, tj. 27–36 milionů lidí. Asi 80 % vzácných nemocí tvoří primárně geneticky podmíněná onemocnění, ale patří sem i četné autoimunitní a infekční nemoci, vzácné formy nádorů, specifické intoxikace a další.
Přehled nejčastějších vzácných onemocnění
- metabolická onemocnění: vrozené poruchy metabolizmu sacharidů (glykogenózy – von Gierkeho nemoc, Pompeho nemoc), lipidů (Gaucherova nemoc, Fabryho nemoc, Niemannova-Pickova nemoc), aminokyselin (fenylketonurie, alkaptonurie, tyrozinemie), mukopolysacharidózy, albinizmus, porfyrie, mitochondriální poruchy, osteopatie a chondropatie
- systémová autoimunitní a revmatologická onemocnění: dermatomyozitida, polymyozitida, sklerodermie, Stillova nemoc, vaskulitidy (temporální arteriitida, polyarteriitis nodosa, Wegenerova granulomatóza), amyloidóza
- kardiovaskulární onemocnění: plicní arteriální hypertenze, arytmogenní dysplazie pravé komory, Brugadův syndrom, syndrom prodlouženého QT intervalu
- endokrinní onemocnění: akromegalie, endogenní Cushingův syndrom, hyperaldosteronizmus, feochromocytom, polyglandulární autoimunitní syndromy, syndromy mnohočetné endokrinní neoplazie, kongenitální adrenální hyperplazie, monogenní diabetes mellitus, genetická obezita (Praderův-Williho syndrom)
- poruchy imunity a autoinflamatorní onemocnění: primární imunodeficity, hereditární angioedém, morbus Behçet, periodické horečky (periodická středomořská horečka, TRAPS – periodický syndrom asociovaný s receptorem pro TNF)
- neuromuskulární onemocnění: periodické svalové paralýzy, Huntingtonova chorea, amyotrofická laterální skleróza, Charcotova-Marieova-Toothova nemoc
Vzácná onemocnění mají často chronický a progredující charakter a jsou obvykle spjata s nepříznivou prognózou. I když je většina z nich nevyléčitelná, vhodná terapie může vést k významnému prodloužení života postižených a zároveň do značné míry zlepšit jeho kvalitu. Předpokladem toho je včasná a správná diagnostika, která je však komplikovaná velkou rozmanitostí vzácných onemocnění, jakož i nedostatkem poznatků o jejich klinických projevech a diagnostických možnostech. I když se první příznaky u řady vzácných onemocnění rozvíjejí již v raném dětství, asi polovina z nich se projeví teprve v dospělosti, přičemž takto postižení pacienti se nejčastěji objevují v ambulancích praktických lékařů nebo všeobecných internistů. Cílem České internistické společnosti (ČIS) České lékařské společnosti J. E. Purkyně je proto za účelem zrychlení a zkvalitnění diagnostického procesu zvyšovat povědomí o jednotlivých vzácných nemocech mezi odbornou i laickou veřejností a zároveň vytvořit síť odborníků z řad internistů, kteří by sloužili jako referenční a koordinační centra pro screening a diagnostiku vzácných onemocnění pro jednotlivé regiony. Další oblastí, která zatím do značné míry unikala pozornosti a v níž by právě všeobecný internista mohl hrát klíčovou roli, je problematika péče o pacienty s pediatrickými vzácnými nemocemi po dosažení dospělosti. K naplnění těchto cílů plánuje ČIS využít spolupráci s dalšími národními internistickými společnostmi, jakož i s Evropskou federací interní medicíny (EFIM) a s její pracovní skupinou pro vzácná onemocnění založenou v roce 2010.
Hyperlipoproteinemie jako vzácná onemocnění
Vzácné hyperlipoproteinemie (HLP) a dyslipidemie (DLP) můžeme rozdělit především podle převažující metabolické poruchy na primární hypercholesterolemie, primární smíšené HLP a primární hypertriglyceridemie. V předloženém sdělení se budeme věnovat především dvěma poruchám, homozygotní familiární hypercholesterolemii (FH) a deficitu lipoproteinové lipázy (LPLD). Dotkneme se i HLP typu III, nazývané někdy dysbetalipoproteinemie. Ostatní vzácné HLP jen stručně uvedeme v tab. 1.

Familiární hyperlipoproteinemie typ I
Familiární hyperlipoproteinemie typ I vzniká jako deficit lipoproteinové lipázy nebo deficit ApoC II, který je kofaktorem nutným k aktivaci tohoto enzymu. Často užívanou zkratkou pro toto onemocnění je LPLD nebo LLD z anglického lipoprotein lipase deficiency. Jedná se o vzácné onemocnění (1 : 1 000 000) charakterizované hyperchylomikronemií a extrémně vysokými hladinami triglyceridů (TG). Cholesterol (C) je zvýšen, ale jen mírně, může být i zcela normální. Jen pro orientaci uvedeme, že hladiny TG se pohybují mezi 10–50 mmol/l (ale mohou být ještě vyšší). Mechanizmus vzniku je schematicky znázorněn na obr. 1. Sérum má skutečně bez přehánění mléčný až smetanový vzhled (obr. 2).


V klinickém obraze dominuje nález abdominálních kolik, u vysokého procenta nemocných lze prokázat pankreatitidu, která může být i fatální. Léčba je svízelná, jedinou možností byla po dlouhá léta velmi přísná dieta. Kazuistická sdělení uvádějí nyní nově možnost využití lomitapidu. Ten snižuje syntézu chylomikronů ve střevě a současně i produkci VLDL v játrech (obr. 3).

Problémem této účinné léčby je riziko hepatopatie, která se může rozvinout po letech léčby (steatofibróza až cirhóza).
Zcela zásadní změnu v terapii pro léčbu by mohla přinést genová terapie. Několik pacientů již genovou terapii (Glybera®) podstoupilo. Výsledky jsou krátkodobě (v řádu měsíců) výborné z hlediska dosažených hladin lipidového a lipoproteinového metabolizmu, posléze mají hodnoty lipidů tendenci ke vzestupu. Pro nemocné je však podstatné, že ani po vzestupu lipidů nedochází k recidivám pankreatitid. Genová terapie je zatím soustředěna do několika center ve světě. Její výsledky musíme dlouhodobě sledovat a ověřovat, protože se jedná o další možný přelom v léčbě poruch tukového metabolizmu.
V každém případě je třeba vnímat LLD jako onemocnění, které může být pro nemocného fatální. Nechceme na tomto místě vyzývat k soutěži, je ale pravda, že v současné době je akutní pankreatitida minimálně stejně závažná (možná s ještě horšími následky i mortalitou) jako infarkt myokardu, který je komplikací hypercholesterolemie. Právě proto, že akutní pankreatitida je někdy prvním projevem LLD, je třeba po LLD aktivně pátrat.
Jaké jsou současné možnosti screeningu LLD? S ohledem na extrémně nízký výskyt v populaci je screening obtížný.
- První možností je pátrat ve skupině nemocných s akutní pankreatitidou, samozřejmě je třeba především provést u nemocného vyšetření základních lipidů, tedy cholesterolu a triglyceridů, při výrazné elevaci triglyceridů (nad 10 mmol/l), potom zejména, pokud je normální nebo jen mírně zvýšený cholesterol, by bylo vhodné vyšetření specialistou na poruchy tukového metabolizmu, při podezření na LLD pak zajistit vyšetření aktivity lipoproteinové lipázy a posléze vyšetření genetické.
- Druhá možnost vyžaduje spolupráci s odděleními klinické biochemie. Pacienti, kteří mají těžkou hypertriglyceridemii (nejméně nad 10 mmol/l) by měli být pozváni k podrobnému klinickému vyšetření a dle jejich stavu a eventuální další biochemické kontroly je možno pokračovat jako v prvním bodě.
- Třetí možností je v zemi, která má propracovaný systém lipidových poraden, které navíc koordinují svoji činnost v rámci projektu MedPed, pověřit tyto poradny, aby provedly reevaluaci všech pacientů se suspektní LLD.
- Čtvrtá, poslední možnost je zvýšit obecné povědomí o LLD u lékařské veřejnosti a doufat, že nemocní budou zachyceni.
V každém případě bychom měli na LLD, jako na vzácné onemocnění s možnou fatální komplikací a nyní s novými možnostmi léčby, myslet. Včasné rozpoznání a léčba mohou zásadně změnit osud těchto nemocných.
Je třeba na toto onemocnění myslet při abdominálních kolikách u dětí a také při pankreatitidách, např. po zahájení hormonální léčby nebo v těhotenství.
Familiární hypercholesterolemie v homozygotní formě a těžká heterozygotní FH
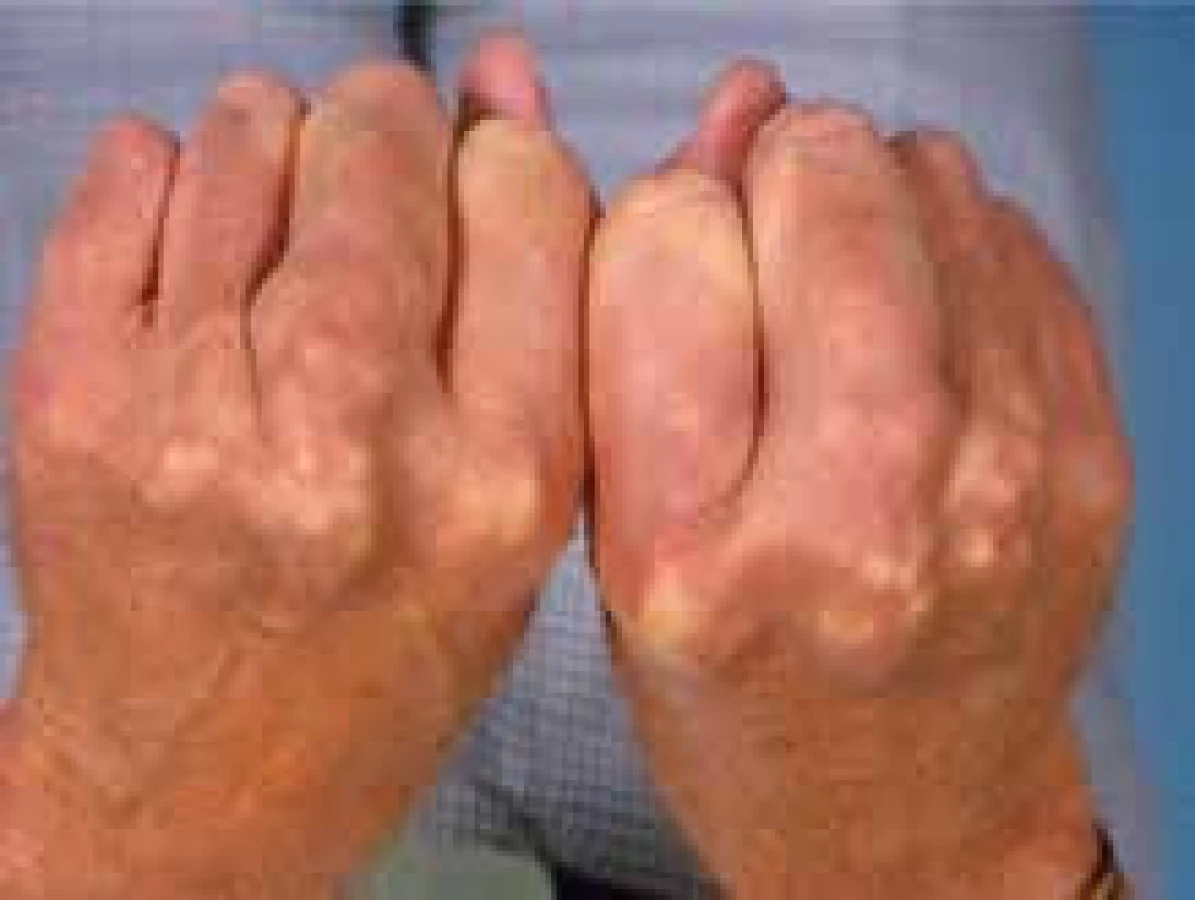
Dalším, velmi významným a potenciálně fatálním vzácným onemocněním z oblasti poruch lipidového metabolizmu je familiární hypercholesterolemie (FH) v homozygotní formě. Její frekvence je mezi 1 : 160 000 až 1 : 1 000 000. Tato čísla platí pro většinu populace světa, v některých zemích (Jižní Afrika, Libanon), v nichž se uplatnil efekt zakladatele rodu (founder effect), je výskyt homozygotů vyšší, pořád však bohatě splňuje kritéria vzácné choroby. V širším slova smyslu je možno za vzácné onemocnění považovat i heterozygotní formu, tzv. severe FH, tedy těžkou heterozygotní FH s mnohými klinickými projevy homozygota (obr. 4, 5, 6).


FH je na jedné straně příkladem, který nám poskytla sama příroda ke studiu vztahů lipidového metabolizmu (především LDL-C) k ateroskleróze a předčasné manifestaci kardiovaskulárních onemocnění. Je ale bohužel také, a to především, nejvýznamnějším klinickým syndromem, který vede k rozvoji ischemické choroby srdeční, prostřednictvím vrozeně zvýšené koncentrace LDL-C.
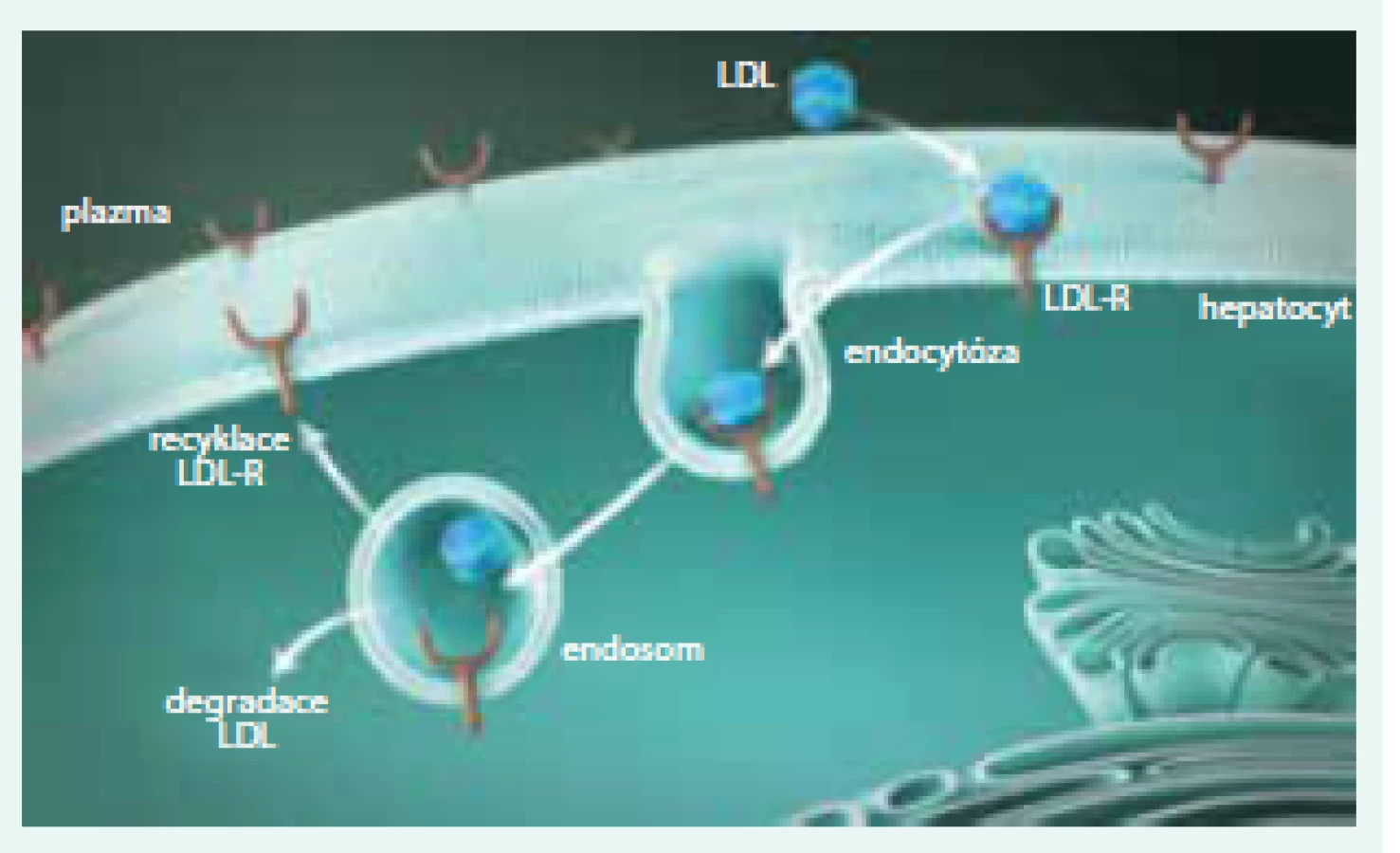
FH je charakterizována (až extrémně) zvýšenými koncentracemi LDL-C. FH byla, a do značné míry stále je, považována za „receptorovou nemoc“. LDL receptor je bílkovina, která je umístěna na povrchu buňky a v zásadě „vystupuje“ z buněčné membrány. Zde je typickou lokalizací LDL receptoru oblast potažené jamky (coated pit), v níž dochází k vlastní interakci s LDL částicí. Komplex LDL receptor + LDL částice je pak internalizován, zavzat do buňky, transportován v „potaženém váčku“ a dále pak dochází, po působení lyzosomů a štěpení, k využití jednotlivých částí. Cholesterol je využit pro potřeby buňky. LDL receptory jsou buď recyklovány a putují na povrch buňky, kde nadále standardně fungují, nebo jsou degradovány v lyzosomech (obr. 4). Je důležité uvést, že k degradaci LDL receptoru přispívá enzym proproteinkonvertáza subtilizin/kexin-9 (PCSK9). Pokud se PCSK9 naváže na LDL receptor, ten zaniká a nevrací se na povrch buňky (obr. 7, 8).

Z výše uvedeného vyplývá, že příčiny FH mohou být v zásadě tři. Klasickou FH je samozřejmě geneticky determinovaný defekt LDL receptoru. Nejsou-li (funkční, či numericky) LDL receptory, dochází k hromadění LDL-C v plazmě s důsledkem předčasné manifestace kardiovaskulárního onemocnění. Koncem 80. let minulého století byli popsáni nemocní s klinicky typickou FH a normálním počtem i funkcí LDL receptorů. Zkoumání těchto pacientů vedlo k odhalení defektu v apolipoproteinu B-100, který je nazýván familiárně defektní ApoB-100, nebo jen FDB. Důsledky z hlediska množství LDL-C v plazmě jsou u FDB podobné jako u FH. Nejnovější výzkumy pak prokázaly, že k obrazu FH může vést i mutace v genu pro PCSK9. Vyšší aktivita PCSK9 snižuje možnost recyklace LDL receptorů na povrch buňky a počet LDL receptorů klesá s příslušným metabolickým efektem.
Diagnostika FH
Protože se tento článek věnuje především homozygotní formě FH a těžkým heterozygotům, z hlediska diagnostiky uvedeme jen to základní: zjednodušené biochemické doporučení a tabulku holandských kritérií pro diagnostiku FH.
Komplexnější vyšetření si zaslouží každý nemocný s celkovým cholesterolem > 7,5 (bez pochyb > 8) mmol/l nebo LDL-C > 5,5 (bez pochyb 6) mmol/l. Je to obrovské zjednodušení, které si jistě zaslouží kritiku (jíž se vůbec nebudeme bránit, je oprávněná).
Podívejme se ale na oficiální diagnostiku FH. Existuje několik skórovacích systémů, pro zájemce uveďme nejpropracovanější kritéria Dutch Lipid Clinic Network (tab. 2).

Pro jistou diagnózu FH musí subjekt dosáhnout více než 8 bodů. Pravděpodobnou diagnózu FH přináší výsledek 6–8 bodů. Jako možnou diagnózu FH označujeme hodnotu 3–5 bodů a naopak ti, kteří dosáhli 0–2 bodů, pravděpodobně FH nemají.
Nejzávažnější klinickou manifestací FH je ICHS. V době „před statiny“ byla mortalita nemocných s FH ve věku 20–40 let stonásobně vyšší než u ostatní populace! ICHS se objevuje u homozygotů již v prvních dekádách a pacienti umírají (pokud nejsou léčeni hypolipidemiky, ale především včasnou revaskularizací a stále častěji též transplantací srdce) do 30 let věku. Jen na okraj bych si dovolil podotknout, že první transplantaci srdce provedl prof. Barnard v Kapském Městě právě u nemocného s FH. U heterozygotů je průběh závažnější u mužů, polovina jich umírá na ICHS do 60 let, na rozdíl od „pouhých“ 15 % žen. U žen dochází k manifestaci ICHS asi o 9 let později než u mužů. K manifestaci ICHS dochází u mužů ve věku 42–46 let, u žen ve věku 51–52 let. Je zajímavé, že pacienti, zejména homozygoti, mají horší prognózu, pokud mají „receptor negativní“ mutaci ve srovnání s nositeli mutace „receptor defektní“. Prognózu zhoršuje především kouření cigaret a někdy hypertenze.
Z klinického hlediska je důležité uvést, že pacient s FH nemá častěji hypertenzi, už vůbec ne diabetes mellitus 2. typu (výskyt DM2T je nižší než ve srovnatelné skupině), bývá spíše štíhlý, nesplňuje kritéria metabolického syndromu a ani nemá hyperurikemii.
Homozygotní forma familiární hypercholesterolemie
Homozygotní forma FH je vzácné onemocnění, které je klinicky nejjednodušeji charakterizováno hladinou cholesterolu > 13 mmol/l, xantomatózou a výrazně předčasnou manifestací KV onemocnění (dokonce i v dětském věku). Notoricky opakovaná (i výše v textu uvedená) frekvence výskytu 1 : 1 000 000 pravděpodobně bude vyšší a bude asi 1 : 160 000–1 : 300 000. I tak pravděpodobně většina čtenářů naší publikace homozygota FH ve své klinické praxi nepotkala ani nepotká. O to důležitější ale je, aby bylo včas vysloveno podezření a nemocný byl odeslán na specializované pracoviště. Kromě cholesterolu > 13 mmol/l je v biochemii zvýšený i LDL-C > 8 mmol/l (obvykle vyšší). Arcus lipoides, xantelazma palpebrarum a šlachové xantomy jsou nesrovnatelně častější než u heterozygotů. Farmakoterapie zahrnuje statiny v maximální dávce v kombinaci s ezetimibem, ale jen zřídka dosáhneme nebo se jen přiblížíme k cílovým hodnotám. Léčbou je pak LDL-aferéza. V současnosti je schválen v EU a USA lomitapid, v USA mipomersen. Transplantace jater (někdy společná s transplantací srdce) je metodou individuální a experimentální.
Naproti tomu zcela novou naději přinášejí dokonce i homozygotům FH nové léky ze skupiny PCSK9 inhibitorů, již schválené evolokumab a alirokumab (Repatha a Praluent), nebo bokozikumab (ve stadiu klinického ověřování). Po léčbě těmito léky se snižuje potřeba LDL-aferéz a pacienti dosahují dříve nepředstavitelných poklesů hladin LDL-C.
Těžký heterozygot FH (severe heterozygous FH)
Definice těžkého heterozygota FH je důležitá, protože tato skupina nemocných je v extrémně vysokém riziku KVO a potřebuje nejintenzivnější terapii. V budoucnosti se z těchto nemocných budou rekrutovat kandidáti tzv. biologické léčby a dalších moderních léčebných postupů. Za kritérium pro diagnózu těžkého heterozygota byla klasicky považována pouze hladina LDL-C 8,0 mmol/l. Holandští autoři tento názor racionálně revidovali a vztáhli „těžkost“ především k rizikovosti rozvoje KVO. Proto dnes do skupiny tzv. severe heterozygous FH zařazujeme i nemocné s FH a kombinací dalších RF KVO, i když hladiny 8,0 mmol/l nedosahují. Při stanovení diagnózy „těžkého heterozygota FH“ je třeba zohlednit také věk a pohlaví nemocného. Pokud jde o terapii, ta kopíruje situaci u homozygotů, užívají se tedy především vysoce dávkované nejúčinnější statiny, případně v kombinaci. Zejména těžcí heterozygoté FH budou léčeni výše uvedenými PCSK9 inhibitory.
HLP typ III (familiární dysbetalipoproteinemie) je vrozenou poruchou metabolizmu lipoproteinů charakterizovanou vysokou hladinou cholesterolu i TG. Podkladem tohoto onemocnění je defekt genu pro apolipoprotein E (ApoE). ApoE je zodpovědný za receptory zprostředkovanou clearance lipoproteinových částic bohatých na TG. Více než 90 % nemocných s HLP III má genotyp E2/E2, zdaleka ne u všech nositelů tohoto genotypu se však HLP III vyvine. K manifestaci HLP III je kromě tohoto polymorfizmu třeba dalšího precipitujícího faktoru. (Při zmínce o polymorfizmu ApoE je třeba uvést, že izoforma E4 je častější u nemocných s kognitivním deficitem i Alzheimerovou chorobou.)
Nejzávažnější klinickou komplikací HLP typu III je předčasná ateroskleróza, která postihuje spíše periferní tepny, nevyhýbá se však ani koronárnímu řečišti. Nemocní s tímto typem hyperlipoproteinemie mohou být postiženi prakticky všemi typy xantomatózy. Léčbu začínáme přísnou dietou, z medikamentů jsou na prvém místě fibráty. Není-li léčba dostatečně efektivní, přidáme k fibrátu statin. Nezapomínáme ani na léčbu eventuální precipitující příčiny, kterou může být dekompenzace diabetes mellitus nebo hypotyreóza.
Závěr
HLP a DLP jsou samozřejmě především vnímány jako onemocnění masového výskytu, významné rizikové faktory KV onemocnění. Je to samozřejmě pravda. Na druhé straně nelze přehlížet, že existují skupiny pacientů, jejichž porucha tukového metabolizmu splňuje kritéria vzácného onemocnění. Jsou to přitom onemocnění, která svému nositeli přinášejí fatální rizika, jako je akutní nekrotizující pankreatitida nebo fatální kardiovaskulární příhoda. S ohledem na to, že léčba těchto onemocnění je možná (jakkoliv nákladná), musíme na tato vzácná onemocnění pomýšlet. Je samozřejmé, že těmto nemocným musí být věnována mimořádná pozornost. Je třeba jim poskytnout péči napříč různými obory a současně se věnovat i široké edukaci medicínské veřejnosti.
Článek vznikal s částečným pouzitim textu Češka R, Votavová L, Schwarzová L et al. Hyperlipoproteinemie jako vzácná onemocnění, který byl uveřejněn v časopise Postgraduální medicína 2016; 18(příl. 2).
Práce byla částečně podpořena granty: AZV 15–28876A, AZV 15–28277A, IAS – Pfizer IGLC Grant. Článek vznikl za částečné podpory projektu ScreenPro FH.
Ing. Lucie Votavová
votavova@interna-cz.eu
ScreenProFH, z.s.,
Praha
www.screenprofh.com
Doručeno do redakce 10. 10. 2016
Přijato po recenzi 2. 11. 2016
Sources
1. Piepoli MF, Hoes AW, Agewall S et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2016; 37(29): 2315–2381. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehw106>.
2. Catapano AL, Graham I, De Backer G et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias: The Task Force for the Management of Dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS)Developed with the special contribution of the European Assocciation for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J 2016. pii: ehw272. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehw272>.
3. Santos RD, Gidding SS, Hegele RA et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes-Endocrinol 2016; 4(10): 850–861. Dostupné z DOI: <http://dx.doi.org/10.1016/S2213–8587(16)30041–9>.
4. Goldstein JL, Hobbs HH, Brown MS. Familial hypercholesterolemia. In: Scriver CR, Beaudet AL(eds) et al. The metabolic and molecular bases of inherited disease. 8th ed. McGraw-Hill: New York 2001 : 2863–2914. ISBN 978–0071363198.
5. Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1992; 1(6): 445–466.
6. Innerarity TL, Weisgraber KH, Arnold KS et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci USA 1987; 84(19): 6919–6923.
7. Fouchier SW, Dallinga-Thie GM, Meijers JC et al. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ Res 2014; 115(6): 552–555. Dostupné z DOI: <http://dx.doi.org/10.1161/CIRCRESAHA.115.304660>.
Labels
Diabetology Endocrinology Internal medicineArticle was published in
Internal Medicine

2016 Issue 11
Most read in this issue
- Dieta při dyslipidemii a metabolickém syndromu
- Hyperlipoproteinemie u dětí
- Hyperlipoproteinemie a dyslipidemie jako vzácná onemocnění: diagnostika a léčba
- Kyselina močová jako rizikový faktor kardiovaskulárních onemocnění