
Hamartom ze zralých kardiomyocytů.
Pitevní kazuistika
Authors:
Šárka Hadravská 1,2; Magdaléna Dubová 1,2; Markéta Miesbauerová 1,2; Petr Mukenšnabl 1; Ondřej Daum 1,2; Alois Mádle 3; Karel Smetana 4
Authors‘ workplace:
Šiklův ústav patologie FN Plzeň, Univerzita Karlova v Praze, Lékařská fakulta v Plzni
1; Bioptická laboratoř, s. r. o., Plzeň
2; II. interní klinika FN Plzeň, Univerzita Karlova v Praze, Lékařská fakulta v Plzni
3; Interní oddělení FN Plzeň, Univerzita Karlova v Praze, Lékařská fakulta v Plzni
4
Published in:
Čes.-slov. Patol., 53, 2017, No. 4, p. 185-187
Category:
Original Article
Overview
Hamartom ze zralých kardiomyocytů (hamartoma of mature cardiac myocytes, HMCM) je vzácná pseudoneoplastická léze myokardu. Autoři popisují případ 39leté Bulharky žijící v České republice, která zemřela v nemocnici na následky ruptury aneuryzmatu lokalizovaného v oblasti arteria communicans anterior a rozsáhlé bronchopneumonie. Při pitvě byl náhodným nálezem šedobílý neopouzdřený neostře ohraničený tumor v přední stěně levé srdeční komory a přilehlé části komorového septa, který mírně prominoval nad rovinu řezu. Mikroskopicky tumor sestával z různých forem dezorganizovaných hypertrofických zralých kardiomyocytů bez vakuolizace cytoplazmy, ložiskově tvořících formace vzhledu „rybí kosti“, s vmezeřenými pruhy vazivové tkáně. Přítomny byly dilatované venuly a silnostěnné větve koronárních arterií. Proliferační aktivita stanovená na základě imunohistochemického průkazu antigenu MIB1 byla nulová. Zánětlivá infiltrace, tuková tkáň ani kalcifikace nebyly v tumoru přítomny.
Klíčová slova:
srdce – hamartom – zralé kardiomyocyty
HMCM je vzácná benigní léze myokardu, charakterizovaná jako lokalizovaná, neopouzdřená, více či méně ohraničená léze tvořená dezorganizovanými hypertrofickými kardiomyocyty bez vakuolizace nebo jen s ojedinělou vakuolizací cytoplazmy, s pruhovitou fibrózou, dilatovanými venulami a silnostěnnými intramurálními větvemi koronárních arterií ve stromatu. Představuje pravděpodobně kongenitální vývojovou anomálii a postihuje nejčastěji levou, méně často pravou srdeční komoru, ojediněle se vyskytuje v pravé srdeční síni a komorovém septu (1-3). HMCM může být klinicky zcela asymptomatický a je náhodným nálezem při jiných vyšetřeních nebo při pitvě, může však způsobit nespecifické změny EKG, poruchy srdečního rytmu, respirační potíže nebo bolesti na hrudi. Svojí lokalizací může vést k obstrukci koronárních tepen, snížení kontraktility myokardu, zúžení srdečních dutin a změnám tvaru srdce. Vzácně může být příčinou náhlé smrti (3-6).
POPIS PŘÍPADU
39letá žena původem z Bulharska byla hospitalizována na neurochirurgické klinice pro subarachnoidální krvácení při ruptuře aneuryzmatu arteria communicans anterior, které bylo léčené konzervativně. Během hospitalizace se její stav zkomplikoval rerupturou aneuryzmatu, vaskulárními spazmy, edémem mozku a bronchopneumonií. Žena po 8 dnech hospitalizace zemřela. Indikována byla klinická pitva, při které bylo nalezeno aneuryzma arteria communicans anterior průměru 8 mm, s rupturou a krvácením do levého frontálního laloku v rozsahu 5,7x2x6 cm a do IV. komory mozkové, a subarachnoidální hematom nad oběma hemisférami supratentoriálně a interhemisferálně. Hmotnost mozku byla 1550 g. V plicích byla ověřena splývající purulentní bronchopneumonie paravertebrálních segmentů v rozsahu obou plicních křídel s fibrinózně purulentní pleuritidou. Náhodným nálezem byl šedobílý neopouzdřený neostře ohraničený elastický útvar rozměrů 4,5x3x3 cm v přední stěně levé srdeční komory, zasahující zčásti do přilehlého komorového septa (obr. 1). Stěna levé srdeční komory byla v tomto místě mírně rozšířená a šedobílá tkáň prominovala nad rovinu řezu. Při prokrájení srdce v lamelách nebyly přítomny jiné patologické změny. Hmotnost srdce byla 350 g.

Retrospektivně byl ve zdravotnické dokumentaci vyhledán záznam echokardiografického vyšetření, které žena absolvovala 12 dní před smrtí v rámci předoperačního vyšetření před plánovaným umělým ukončením gravidity na vlastní žádost. Změny EKG křivky byly interpretovány jako mírná hypertrofie anteroseptální stěny levé srdeční komory.
HISTOPATOLOGICKÉ VYŠETŘENÍ
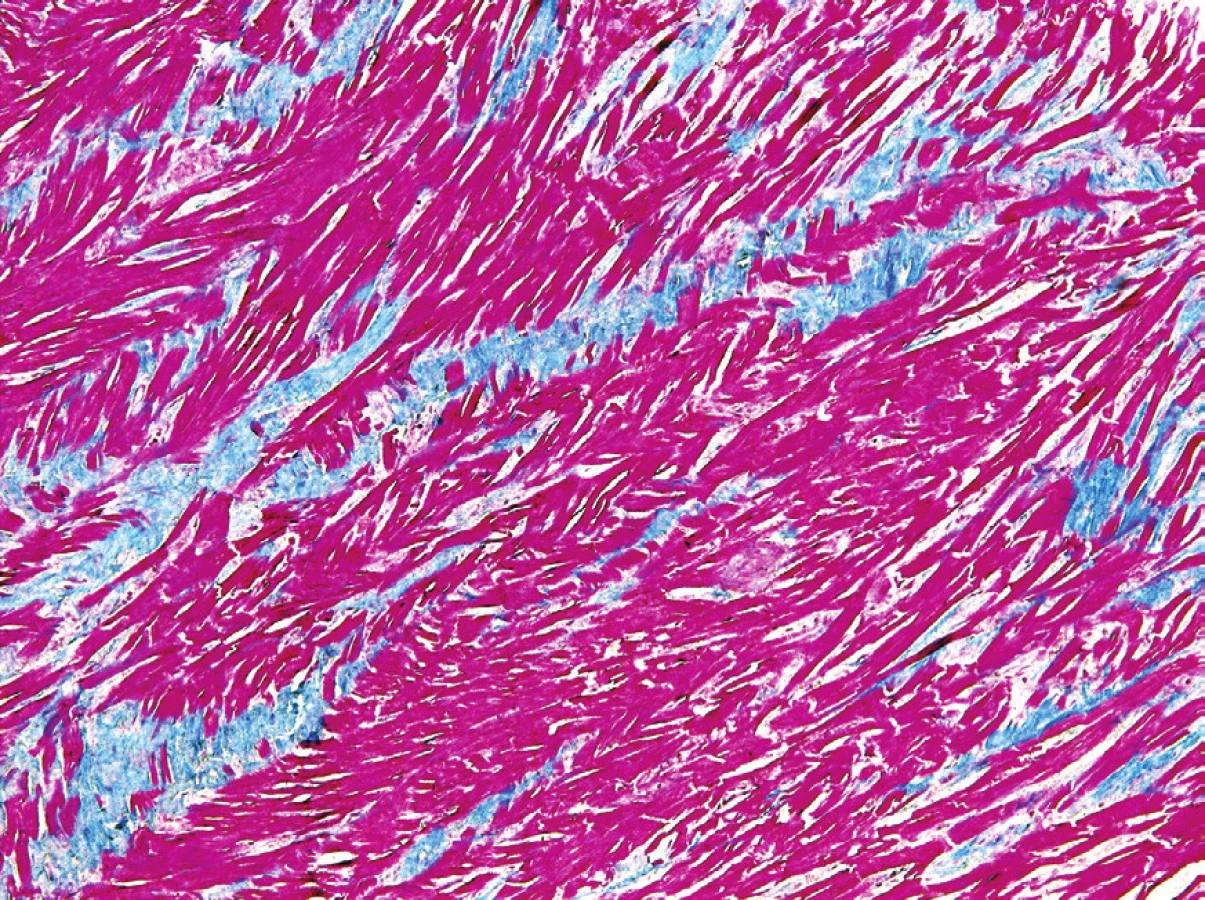
Mikroskopicky byl útvar tvořen dezorganizovanými pruhy hypertrofických zralých kardiomyocytů bez vakuolizace cytoplazmy (obr. 2). Dezorganizace měla nahodilý ráz, místy tvořily kardiomyocyty struktury vzhledu „rybí kosti“ (obr. 3). Jádra kardiomyocytů byla objemná, nepravidelná, místy až bizarního tvaru (obr. 4). Mezi svazky kardiomyocytů byly přítomny pruhy vazivové tkáně (obr. 5), ojedinělé dilatované silnostěnné větve koronárních arterií (obr. 6) a mnohočetné dilatované venuly, jejichž výstelka byla pozitivní v imunohistochemickém průkazu faktoru VIII (obr. 7). Imunohistochemický průkaz antigenu MIB1 ke stanovení proliferační aktivity byl v kardiomyocytech negativní. V lézi nebyla zastižena tuková tkáň, kalcifikace ani zánětlivá infiltrace. Okolní myokard měl normální strukturu. Na základě těchto nálezů byla stanovena diagnóza HMCM.





DISKUZE
HMCM je vzácná benigní pomalu rostoucí pseudoneoplastická léze myokardu, která byla popsána poprvé v r. 1988 (7). Doposud bylo dle našich informací publikováno ve světové literatuře pouze 22 případů HMCM, z toho sedm bylo diagnostikovaných až při pitvě (5). Ostatní případy byly diagnostikovány v rámci operace či bioptického vyšetření (4, 5).
HMCM představuje zřejmě kongenitální vývojovou anomálii postihující nejčastěji stěnu levé srdeční komory (63-90 %), méně často pravé srdeční komory (15 %), a může se vyskytnout i v pravé srdeční síni (8, 9) nebo v komorovém septu (1,2,10,11). V levé srdeční síni nebyl dosud popsán. Většinou je solitární, může však být i mnohotný (8). Vyskytuje se v dětském i dospělém věku, s vyšší prevalencí u mužů (4,5).
HMCM může být klinicky zcela asymptomatický a je pak zcela náhodným nálezem při vyšetření z jiných příčin nebo při pitvě (1,7,9). Někdy může způsobit nespecifické změny EKG (1,8), poruchy srdečního rytmu (10,12), respirační potíže (1), nebo bolesti na hrudi. Svojí lokalizací může vést k obstrukci koronárních tepen, snížení kontraktility myokardu, zúžení srdečních komor nebo síní a změnám tvaru srdce. Vzácně může být příčinou náhlé smrti (1,8).
Mikroskopicky je HMCM charakterizován jako lokalizovaná, neopouzdřená, ale ohraničená léze tvořená dezorganizovanými hypertrofickými zralými kardiomyocyty bez vakuolizace nebo jen s ojedinělou vakuolizací cytoplazmy, s pruhovitou fibrózou a dilatovanými venulami a silnostěnnými větvemi koronárními arterií v intersticiu, přítomna může být i tuková tkáň (3,5,8). Růst je pomalý, často se, jako v našem případě, imunohistochemicky nepodaří prokázat žádnou proliferaci buněk (8). Dezorganizace kardiomyocytů může být nahodilá, ale mohou být vytvořeny i vírovité struktury nebo formace vzhledu „rybí kosti“(1).
Histopatologická diferenciální diagnóza zahrnuje především rhabdomyom, který makroskopicky HMCM připomíná. Vyskytuje se v dětském věku, bývá mnohotný a sestává z nezralých kardiomyocytů obsahujících početné vakuoly glykogenu (13,14). Rhabdomyom je úzce asociován s tuberózní sklerózou a má tendenci ke spontánní regresi v průběhu dětského věku (15).
Hypertrofická kardiomyopatie se makroskopicky prezentuje jako lokalizované a většinou asymetrické ztluštění myokardu, zejména komorového septa (2,6,10). Netvoří ohraničenou lézi, postižená část myokardu je makroskopicky barevně a strukturálně shodná s okolní svalovinou (4) a dezorganizované kardiomyocyty se prolínají s okolním myokardem. Odlišení těchto dvou lézí z malých biopsií je pro histologickou podobnost velmi obtížné (1).
Fibrom srdce je tvořen vazivovou tkání s příměsí elastických vláken, v jejíž periferii mohou být zavzaty kardiomyocyty (16). Na rozdíl od HMCM může kalcifikovat (17).
Histiocytoidní (onkocytární) kardiomyopatie je charakterizovaná subendokardiálními noduly tvořenými kardiomyocyty s onkocytickou přeměnou cytoplazmy. Obvykle se tato léze vyskytuje u dívek (4,18).
ZÁVĚR
V pitevní kazuistice prezentujeme náhodný nález vzácného pseudotumoru srdce u 39leté ženy, která zemřela v důsledku ruptury aneuryzmatu arteria communicans anterior. EKG vyšetření provedené 12 dní před smrtí prokázalo pouze nespecifické změny, které byly interpretovány jako mírná ložisková hypertrofie stěny levé srdeční komory.
PODĚKOVÁNÍ
Práce byla podpořena grantem MŠMT SVV-2017 – 260 391. The work was supported by the Ministry of Education, Youth and Sports grant SVV-2017 – 260 391.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
MUDr. Magdaléna Dubová
Šiklův ústav patologie LF UK a
FN Plzeň
Edvarda Beneše 1128/13,
305 99 Plzeň
tel.: 377 402 089
fax.: 377 402 634
e-mail: dubovam@fnplzen.cz
Sources
1. Fealey ME, Edwards WD, Miller DV, Menon SC, Dearani JA. Hamartomas of mature cardiac myocytes: report of 7 new cases and review of literature. Hum Pathol 2008; 39(7): 1064-1071.
2. Miller DV, Tazelaar HD. Cardiovascular pseudoneoplasms. Arch Pathol Lab Med 2010; 134(3): 362-368.
3. Zhang F, Yin N, Yin B, Xu S, Yang Y. Giant right atrial cystic hamartoma: a case report and literature review. BMJ Case Rep 2009. pii: bcr02.2009.1587.
4. Galeone A, Validire P, Gayet JB, Laborde F. Hamartoma of mature cardiac myocytes of the pulmonary infundibulum. Interact Cardiovasc Thorac Surg 2009; 9(6): 1029-1031.
5. Raffa GM, Malvindi PG, Settepani F, et al. Hamartoma of mature cardiac myocytes in adults and young: case report and literature review. Int J Cardiol 2013; 163(2): e28-30.
6. Menon SC, Miller DV, Cabalka AK, Hagler DJ. Hamartomas of mature cardiac myocytes. Eur J Echocardiogr 2008; 9(6): 835-839.
7. Tanimura A, Kato M, Morimatsu M. Cardiac hamartoma. A case report. Acta Pathol Jpn 1988; 38(11): 1481-1484.
8. Burke AP, Ribe JK, Bajaj AK, Edwards WD, Farb A, Virmani R. Hamartoma of mature cardiac myocytes. Hum Pathol 1998; 29(9): 904-909.
9. Movahedi N, Boroumand MA, Sotoudeh Anvari M, Yazdanifard P. Mature cardiac myocyte hamartoma in the right atrium. Asian Cardiovasc Thorac Ann 2008; 16(5): e47-48.
10. Gilman G, Wright RS, Glockner JF, et al. Ventricular septal hamartoma mimicking hypertrophic cardiomyopathy in a 41-year-old woman presenting with paroxysmal supraventricular tachycardia. J Am Soc Echocardiogr 2005; 18(3): 272-274.
11. Sturtz CL, Abt AB, Leuenberger UA, Damiano R. Hamartoma of mature cardiac myocytes: a case report. Mod Pathol 1998; 11(5): 496-499.
12. Dinh MH, Galvin JM, Aretz TH, Torchiana DF. Left ventricular hamartoma associated with ventricular tachycardia. Ann Thorac Surg 2001; 71(5): 1673-1675.
13. Fenoglio JJ, Jr., M. CAllister HA J, Ferrans VJ. Cardiac rhabdomyoma: a clinicopathologic and electron microscopic study. Am J Cardiol 1976; 38(2): 241-251.
14. Burke AP, Virmani R. Cardiac rhabdomyoma: a clinicopathologic study. Mod Pathol 1991; 4(1): 70-74.
15. de Silva DC, Johnston AW, Dean JC. Tuberous sclerosis--an unusual cause of seizures in an 18 year old. Scott Med J 1994; 39(1): 19-20.
16. Feldman PS, Meyer MW. Fibroelastic hamartoma (fibroma) of the heart. Cancer 1976; 38(1): 314-323.
17. Burke AP, Rosado-de-Christenson M, Templeton PA, Virmani R. Cardiac fibroma: clinicopathologic correlates and surgical treatment. J Thorac Cardiovasc Surg 1994; 108(5): 862-870.
18. Shehata BM, Patterson K, Thomas JE, Scala-Barnett D, Dasu S, Robinson HB. Histiocytoid cardiomyopathy: three new cases and a review of the literature. Pediatr Dev Pathol 1998; 1(1): 56-69.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2017 Issue 4
Most read in this issue
- Co nového v Ewing-like family aneb malobuněčné/kulatobuněčné sarkomy měkkých tkání a kostí s rearanží genů CIC a BCOR. Přehled problematiky a naše prvotní zkušenosti
- Molekulární patologie plicních karcinomů pro rutinní praxi – update 2017
- Molekulární mechanizmy primární a sekundární rezistence, molekulárně-genetické znaky a vlastnosti KIT/PDGFRA nemutovaných GIST
-
Hamartom ze zralých kardiomyocytů.
Pitevní kazuistika