
Gorlinův-Goltzův syndrom – popis případu a přehled literatury
Authors:
M. Důra 1,2; J. Schmittová 4; D. Polendová 5; D. Kacerovská 2,3
Authors‘ workplace:
Dermatovenerologická klinika 1. LF UK a VFN, Praha
1; Bioptická laboratoř s. r. o., Plzeň
2; Šiklův ústav patologie LF UK a FN Plzeň
3; DERMACENTRUM Schmittová s. r. o., Strakonice
4; Ústav lékařské genetiky LF UK a FN Plzeň
5
Published in:
Čes-slov Derm, 100, 2025, No. 5, p. 197-201
Category:
Case Reports
Overview
Gorlinův-Goltzův syndrom je vzácné, geneticky podmíněné onemocnění. Mezi typické projevy tohoto onemocnění patří mnohočetné bazocelulární karcinomy vznikající v mladém věku (i v dětství), čelistní cysty, palmoplantární jamky, skeletální abnormality a další vývojové vady. Gorlinův-Goltzův syndrom je monogenní onemocnění, zapříčiněné nejčastěji mutací v genu PTCH1, méně často v genech PTCH2 či SUFU. U pacientů nesoucích mutace v těchto genech je zvýšené riziko vzniku primárně mozkových nádorů, zejména meduloblastomu, a kardiálních fibromů. Pacienti s Gorlinovým-Goltzovým syndromem by měli být pravidelně sledováni dermatologem, stomatologem a kardiologem. Tato práce prezentuje případ 16letého pacienta s Gorlinovým-Goltzovým syndromem s výskytem vícečetných bazocelulárních karcinomů a čelistních cyst. Diagnóza byla potvrzena molekulárně-genetickým testováním, které pro kázalo heterozygotickou mutaci genu PTCH1 (708G > A) jak u pacienta, tak u jeho matky. Autoři předkládají přehled současných poznatků o tomto onemocnění.
Klíčová slova:
bazocelulární karcinom – Gorlinův-Goltzův syndrom – bazaloidní folikulární hamartom – PTCH1 mutace – vismodegib
ÚVOD
Gorlinův-Goltzův syndrom je vzácné, geneticky podmíněné onemocnění. Diagnóza tohoto onemocnění je v častých případech stanovena dermatologem, a to vzhledem k výskytu bazocelulárních karcinomů u pacientů zpravidla mladších 20 let. Prezentován je případ pacienta s Gorlinovým-Goltzovým syndromem a připojen je souhrn problematiky.
KLINICKÝ PŘÍPAD
Pacient, t. č. 16letý muž, byl odeslán ke kožnímu vyšetření dětským obvodním lékařem pro výskyt mnohočetných fibrotických výrůstků. Dle anamnézy od útlého věku byla pacientovi odstraňována vícečetná milia, epidermoidní cysty a fibromy obličeje, krku a paží.
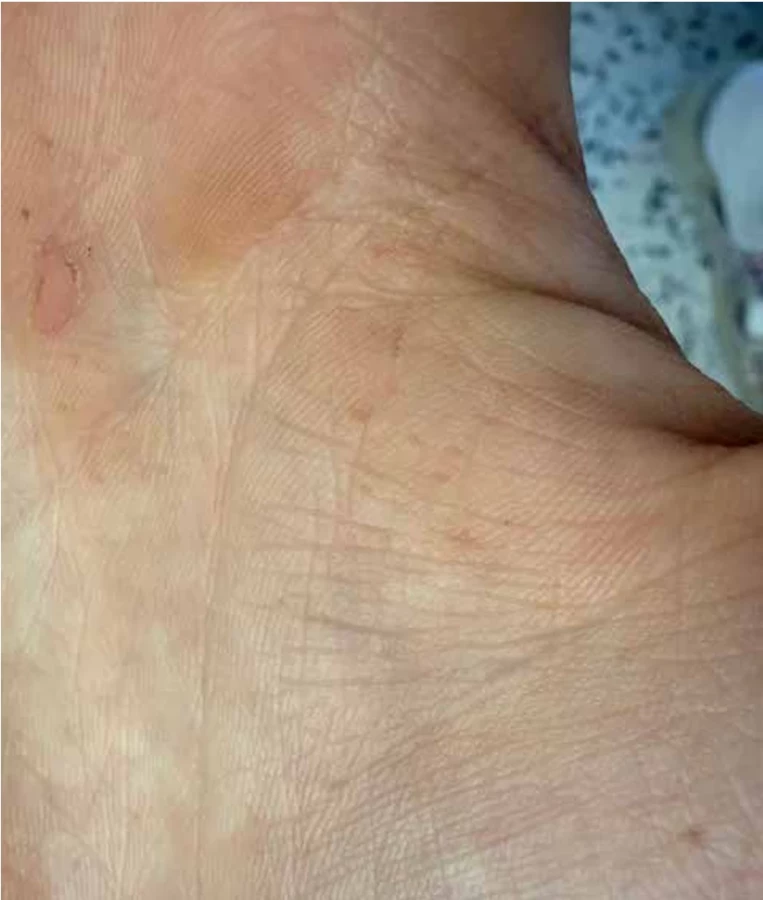
V podbřišku měl pacient několik pigmentací typu café-au-lait skvrn. Klinicky byl pacient vysokého vzrůstu (výšky 187 cm), obvod hlavy činil 59 cm (a byl tedy na 95. percentilu), obličej byl nápadný prominujícím čelem, širokým kořenem nosu, hypertelorismem a nízce posazenými ušními boltci (obr. 1). Na dlaních byly zřejmé vícečetné palmární jamky (obr. 2).

Provedena byla probatorní excize ze suspektního projevu v oblasti pravého obočí, histopatologicky byl verifikován mikronodulární bazocelulární karcinom (BCC) a patologem byla vyjádřena suspekce na Gorlinův-Goltzův syndrom vzhledem k nízkému věku pacienta. V krátkém časovém odstupu byl pacientovi odstraněn trichoblastom obličeje a další dva BCC na trupu (nad levou lopatkou a pod pravou klíční kostí). Posledně excidovaný BCC s přítomností pigmentu pod pravou klíční kostí dokumentují obrázky 3a a 3b.

Pacient je též dispenzarizován stomatologem pro poruchu dentice, konkrétně proretinované zuby a mnohočetné odontogenní cysty. Tyto cysty dokumentuje rentgenový snímek čelistí (obr. 4). Dvakrát byly cysty mandibuly stomatochirurgicky resekovány. Anamnesticky byl pacient vyšetřován neurologem již ve 3 měsících věku pro makrocefalii, následně byl vyřazen z evidence.

Matka pacienta, věku 40 let, dochází též na pravidelné dermatologické kontroly pro výskyt vícečetných BCC, je taktéž poměrně vysokého vzrůstu (výšky 183 cm) a v minulosti prodělala opakované exstirpace čelistních cyst. I u ní jsou přítomny vícečetné palmární jamky včetně bočních partií prstů (obr. 5a, 5b).

U pacientových prarodičů z matčiny strany nebyly známy kožní či stomatologické příznaky, děda byl též vysokého vzrůstu (190 cm). Pacientova sestra, věku 23 let, je zdráva. Otec pacienta je též zdráv.
Vzhledem k výše popsaným příznakům a rodinné anamnéze bylo u pacienta vyjádřeno podezření na diagnózu Gorlinova-Goltzova syndromu. Následné genetické vyšetření potvrdilo heterozygotickou patogenní mutaci genu PTCH1 v lokusu 708G > A, čímž byla suspekce na tento syndrom potvrzena. Stejná genetická aberace byla následně potvrzena u matky pacienta. Genetickému vyšetření se též podrobila babička a sestra pacienta. U obou byla tato genetická aberace vyloučena.
Pacient je nadále sledován dermatologem, stomatologem a kardiologem. Histopatologicky byl dosud potvrzen nodulární BCC, mikronodulární BCC a trichoblastom. Provedená magnetická rezonance mozku vyloučila primární mozkový nádor.
DISKUSE
Gorlinův-Goltzův syndrom je vzácné, geneticky podmíněné multisystémové onemocnění, zvané též syndromem névoidních bazocelulárních karcinomů. Typickou kožní manifestací je vznik mnohočetných bazocelulárních karcinomů (BCC) v mladém věku.
Tento syndrom popsali v roce 1960 Robert J. Gorlin a Robert W. Goltz [4]. Příčinou jeho vzniku je zárodečná mutace v jednom z genů v rámci signální kaskády Sonig Hedgehog (SHH), zodpovídající za diferenciaci buňky a fyziologický embryonální vývoj. Kauzální geny byly dosud identifikovány tři: PTCH1, méně často PTCH2 a SUFU. Jedná se o tumor-supresorické geny, jejichž inaktivační mutace má za následek up-regulaci signální kaskády SHH. Dědičnost je autozomálně dominantní. Onemocnění má vysokou penetranci s variabilní expresivitou. De novo mutace vzniká asi u 50 % pacientů. U mužů a žen se vyskytuje ve stejné míře. Odhadovaná prevalence je v rozmezí 1 : 57 000 až 1 : 256 000. Poněkud vzácnější je tento syndrom u asijské a afroamerické rasy.
Jak již bylo zmíněno výše, nejčastější gen zodpovědný za Gorlinův-Goltzův syndrom je PTCH1. Molekulární biologie vzniku nádorů u těchto pacientů odpovídá tzv. Knudsonově hypotéze dvou zásahů. Zárodečná mutace genu PTCH1 na jedné alele je u 90 % hereditárních BCC doplněna ztrátou heterozygozity (LOH) v daném lokusu. Vývojové a skeletální abnormality jsou způsobeny zřejmě spíše v důsledku haploinsuficience genu PTCH1. Spektrum mutací nalézaných v genu PTCH1 je široké. Delece, inzerce, nonsense, missense a sestřihové mutace byly identifikovány po celé délce genu, nicméně nejčastěji se nacházejí ve dvou velkých extracelulárnícha jedné velké intracelulární smyčce. Mutační analýza genu PTCH1 je prováděna na jednom ze spoluautorských pracovišť (Bioptická laboratoř s. r. o.).
Mnohočetné BCC se u těchto pacientů tvoří již před 20. rokem života. Predilekčně se vyskytují na kůži chronicky osvětlené, mohou však navíc vznikat i mimo tyto lokality, včetně genitálu. Nové BCC vznikají během celého života a jejich celkový počet může dosahovat i několika stovek. Histopatologicky se u pacientů mohou vyskytnout prakticky všechny podtypy nízce i vysoce rizikových BCC, signifikantně častěji se vyskytuje infundibulocystický typ [2]. Histopatologicky však nelze rozlišit sporadický BCC od léze asociované s Gorlinovým--Goltzovým syndromem.
Kromě BCC se u pacientů ve vyšší míře vyskytuje i tzv. bazaloidní folikulární hamartom (BFH). BFH je většinou drobný benigní kožní nádor s folikulární diferenciací. Recentní studie prokázala poměrně vysokou prevalenci BFH u pacientů s Gorlinovým-Goltzovým syndromem (24 %) [1]. Mnohočetný výskyt BFH u jednoho pacienta tedy může upozornit na možnou diagnózu Gorlinova-Goltzova syndromu. Vzhledem k tomu, že BFH vykazuje analogické genové aberace jako BCC, autoři výše uvedené studie předkládají teorii BFH jakožto možného prekurzoru BCC.
Dalšími kožními manifestacemi jsou palmoplantární jamky (často přítomné již od dětství), mnohočetná milia, epidermoidní cysty a perzistence interdigitálních blan nohou a rukou. Pacienti s tímto syndromem mají vysoký vzrůst a typické rysy obličeje – makrocefalii, hypertelorismus, široký kořen nosu a prominující čelo a mandibulu.
Nejčastějším mimokožním příznakem jsou mnohočetné odontogenní keratocysty, častěji lokalizované v oblasti mandibuly, zjištěné až u 90 % pacientů [7]. Mohou být zcela asymptomatické, případně jsou do provázeny poruchami dentice či patologickými frakturami.
Mezi další extrakutánní příznaky patří kostní abnormality žeber či obratlů, polydaktylie či syndaktylie, pectus excavatum či pectus carinatum, kardiální a ovariální fibromy, mezenteriální lymfatické cysty, pleurální cysty, rozštěpové vady, oční abnormality (např. katarakta, mikroftalmie), kalcifikace falx cerebri před 20. rokem života, u mužů pak hypogonadismus a gynekomastie. Asi 5 % pacientů má poruchu intelektu. Výčet mimokožních projevů však není kompletní, popsána byla řada dalších asociovaných abnormalit. V některých případech mohou být příznaky Gorlinova-Goltzova syndromu vyjádřeny již prenatálně, zejména makrocefalií.
U malého počtu pacientů s Gorlinovým-Goltzovým syndromem se vyvíjí meduloblastom v dětském věku, který je však cca 20krát častější u pacientů nesoucích mutaci genu SUFU [3]. Vyšší je i riziko vzniku meningeomu.
Vypracována byla diagnostická kritéria pro stanovení diagnózy Gorlinova-Goltzova syndromu. Pro diagnózu je nutné splnění dvou hlavních kritérií či jednoho hlavního kritéria a dvou vedlejších kritérií. Jejich souhrn udává tabulka 1. V literatuře se výčet kritérií objevuje v několika drobných modifikacích. Z kritérií je zřejmé, že genetické vyšetření není pro stanovení diagnózy ne zbytně nutné.

jednoho hlavního kritéria a dvou vedlejších kritérií
Pacienty s Gorlinovým-Goltzovým syndromem je nutno pravidelně sledovat v dermatologické ambulanci, dle frekvence vzniku nových projevů v horizontu cca 3 měsíců. Při klinickém vyšetření je nezbytné neopomenout oblast kštice a oblasti kožního povrchu slunci permanentně skryté. Důležitá je ochrana před sluncem a sebekontrola.
Nedílnou součástí komplexní péče jsou pravidelné stomatologické kontroly včetně rentgenologického vyšetření. U dětí útlého věku je ke zvážení též MRI vyšetření mozku k vyloučení meduloblastomu, zejména u těch, u kterých byla zjištěna mutace genu SUFU. V indikovaných případech je na místě konzultace klinického genetika, např. v rámci prekoncepční diagnostiky.
Terapeutickými možnostmi nízce rizikových BCC je chirurgická excize, laseroterapie, kryodestrukce, imunomodulační terapie imiquimodem či fotodynamická terapie. U vysoce rizikových BCC je metodou volby kompletní chirurgická excize. Ve starší literatuře je zmiňována systémová terapie retinoidy v preventivním či léčebném podání [5].
U pokročilých, inoperabilních či metastazujících BCC je lékem první volby vismodegib, který vykázal vysokou účinnost a poměrně rychlý nástup účinku. Vismodegib je perorální cílený lék blokující signální kaskádu Sonig Hedgehog v místě receptoru SMO. Pro zahájení léčby není analýza prediktivních markerů nutná. Vismodegib se užívá v dávce 150 mg 1krát denně. Délka užívání není stanovena, avšak vzhledem k jeho poměrně častým nežádoucím účinkům je obvyklou praxí přerušení či vysazení léčby po několika měsících. Úhrada tohoto léku není ze strany SÚKL stanovena, pro zahájení a úhradu léčby je tedy nutné schválení revizním lékařem (informace k 10/2025). Mezi nejčastější komplikace léčby patří výpad vlasů a chlupů, křeče, dysgeuzie (poruchy chuti), nechutenství a hubnutí.
V dalších liniích léčby se dle dosud omezených dat může uplatňovat imunoterapie inhibitory kontrolních bodů imunity namířených proti receptoru PD-1, konkrétně pembrolizumab a cemiplimab [6].
Vzhledem ke zvýšené senzitivitě kůže pacientů s Gorlinovým-Goltzovým syndromem k záření je obecně paliativní radioterapie kontraindikována.
V diferenciální diagnóze Gorlinova-Goltzova syndromu stojí zejména další genodermatózy s výskytem mnohočetných BCC v mladistvém věku (Bazexův-Du préův-Christolův syndrom, Rombo syndrom či xeroderma pigmentosum), dále těžké chronické solární poškození kůže.
Sources
1. BARBIEUX, S., JOUENNE, F., MACHET, M.C. et al. Re-evaluation of the concept of basaloid follicular hamartoma associated with naevoid basal cell carcinoma syndrome: a morphological, immunohistochemical and molecular study. Pathology, 2025, 57(1), p. 49–56.EDIČNÍ PLÁN
2. CALONJE, E., BRENN, T., LAZAR, A.J. et al. McKee‘s Pathology of the Skin. 5th Edition. Amsterdam: Elsevier/Saunders, 2019; 2 vol., p. 1186–1187. ISBN 978-0-7020-6983-3.
3. van DAL, M., MARTENS-DE KEMP, S.R., MOOYAART, A.L., VOOGT, W., WAKKEE, M., DAMMAN, J. Clinicopathological and molecular spectrum of patients with germline SUFU mutations: A case series. J Cutan Pathol., 2024, 51(12), p. 980–986.
4. GORLIN, R.J., GOLTZ, R.W. Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome. N Engl J Med., 1960, 262, p. 908–912.
5. NECHVÁTALOVÁ, H., PIROCHTOVÁ, K. Syndrom nevoidních bazaliomů a léčba retinoidy. Čes-slov Derm, 2000, 75 (6), p. 280–283.
6. RESCHKE, R., RICHTER, J., ENK, A. H., HASSEL, J. C. Use of anti-PD1 blockade after Hedgehog inhibitors or as first-line therapy for Gorlin syndrome. JAMA Dermatol, 2025, 161 (1), p. 104–105.
7. SPADARI, F., PULICARI, F., PELLEGRINI, M., SCRIBANTE, A., GARAGIOLA, U. Multidisciplinary approach to Gorlin-Goltz syndrome: from diagnosis to surgical treatment of jawbones. Maxillofac Plast Reconstr Surg., 2022, 44(1), p. 25.
Labels
Dermatology & STDs Paediatric dermatology & STDsArticle was published in
Czech-Slovak Dermatology

2025 Issue 5
Most read in this issue
- Gorlin-Goltz Syndrome – Case Report and Review of the Literature
- Hand Eczema Part II: Contact Allergies and Contact Allergens
- Erythematosquamous Exanthema with Blisters Showing Hypopyon Sign