
Ovariální cévní malformace – klinická forma prezentace Cowdenova syndromu
Authors:
Jana Pavlacká; Michal Felsinger; Luboš Minář
Authors‘ workplace:
Gynekologicko-porodnická klinika LF MU a FN Brno
Published in:
Ceska Gynekol 2025; 90(5): 388-394
Category:
Case Report
doi:
https://doi.org/10.48095/cccg2025388
Overview
Cíl: Popis případu dívky, u které byla diagnostikována cévní malformace levého ovaria a geneticky potvrzen Cowdenův syndrom. Kazuistika: Pacientka ve věku 10 let byla odeslána na gynekologii pro náhodný nález patologické rezistence v oblasti levých adnex. Klinicky u dívky nebyly gynekologické potíže, ale docházelo k otoku pravého kolene při fyzické zátěži. Dominujícím nálezem při zobrazovacích vyšetřeních byla venolymfatická malformace vycházející z musculus vastus medialis detekovaná magnetickou rezonancí. Po punkci ložiska bylo z bioptického vzorku provedeno genetické vyšetření, které prokázalo mutaci PTEN genu v heterozygotní formě. Tím byl na molekulární úrovni prokázán Cowdenův syndrom. Při vyšetření magnetickou rezonancí byla současně popsána heterogenní expanze v oblasti levého ovaria. Na základě stagingu byla indikována diagnostická laparoskopie s laváží. Peroperačně byly zjištěny mnohočetné adheze v malé pánvi a tumor vycházející z levého ovaria. Byla provedena parciální resekce levého ovaria, peroperačně z kryobiopsie potvrzena cévní malformace, bez doprovodného nálezu tumoru. Z definitivního histologického vyšetření resekátu levého ovaria byl prokázán výskyt cévních malformací, což klinicky odpovídá možným příznakům Cowdenova syndromu. Závěr: Cowdenův syndrom je vzácné genetické onemocnění, jehož diagnostika vychází z klinických projevů, zobrazovacích vyšetření a následné genetické testace. Sledování pacientek se řídí doporučením National Comprehensive Cancer Network a vyžaduje multidisciplinární přístup.
Klíčová slova:
Cowdenův syndrom – PTEN hamartoma tumor syndrom – nádory prsu a endometria – cévní malformace
Úvod
Cowdenův syndrom (CS) je geneticky podmíněné onemocnění, které je součástí skupiny poruch známých jako PTEN hamartoma tumor syndrom (PHTS). Byl poprvé popsán Lloydem a Dennisem v roce 1963 ve zprávě o 20leté pacientce jménem Rachel Cowden, podle níž byl syndrom následně pojmenován [1]. Jednalo se o zjevně nový klinický obraz charakterizovaný výskytem mnohočetných hamartomů, neobvyklými kožními a obličejovými nálezy, abnormalitami centrálního nervového systému a fibrocystickou mastopatií. V dnešním kontextu jeho charakteristika zahrnuje predispozici k benigním a maligním nádorům, vč. nádorů prsu, štítné žlázy a endometria [2]. Hlavní příčinou CS jsou mutace v genu PTEN, což je tumor supresorový gen. Ten pomáhá regulovat buněčný růst a brání vzniku rakoviny tím, že zpomaluje buněčné dělení a podporuje apoptózu. Případná mutace PTEN může vést k nekontrolovanému dělení buněk a ke vzniku nádorů [3].
Cowdenův syndrom vykazuje autozomálně dominantní typ dědičnosti. Typické genetické abnormality se vyskytují většinou v exonech genu PTEN na chromozomu 10q 22–23, přičemž bez předchozího genetického vyšetření může být diagnostika obtížná [4]. Prevalence CS se celosvětově odhaduje na 1/200 000 [5].
Klinické projevy
Nejčastějšími prezentujícími se lézemi u CS jsou mukokutánní léze, jako jsou orální papilomy, trichilemomy a akrální keratózy, které se vyskytují u 99 % pacientů do třetí dekády života. Projevy onemocnění se mění s věkem. U dospělých se vyskytují typické příznaky CS, děti mají často makrocefalii a poruchy autistického spektra [6].
Klinické projevy CS se mohou výrazně lišit mezi jednotlivými pacienty, což ztěžuje diagnostiku. V roce 2009 provedli Pilarski et al. přehled literatury s cílem aktualizovat klinické rysy spojené s mutacemi PTEN a související klinické syndromy [7]. Národní onkologická síť (NCCN – National Comprehensive Cancer Network) tato kritéria přijala a každý rok je aktualizuje. V tab. 1 je přehled osmi hlavních a deseti vedlejších klinických znaků a symptomů, které jsou typické pro CS.
Potvrzení klinické diagnózy CS je stanoveno, pokud jedinec splňuje kritéria definovaná v tab. 2. NCCN nespecifikuje klinická kritéria u dětí, ta stanovili Tan et al. v roce 2011 [8,9] a jsou také zmíněna v tab. 3. U dětí je nezbytným kritériem pro stanovení diagnózy přítomnost makrocefalie (okcipitofrontální obvod > 2 směrodatné odchylky nad populačním průměrem).
Neurologické příznaky (autizmus a vývojové opoždění) a příznaky kožní (lipomy, orální papilomy) představovaly extrémně časté sekundární projevy CS; jeden příznak nebo oba byly vyjádřeny u 100 % pacientů se zárodečnou mutací PTEN. Avšak vzhledem k tomu, že dermatologické rysy mohou být často přehlédnuty, je pravděpodobné, že jsou u pacientů při prvních projevech v dětství stejně důležité i méně časté rysy, jako jsou cévní malformace, gastrointestinální polypy nebo struma štítné žlázy.
Kůže je hlavním postiženým orgánem, nejčastěji lézemi ve formě hamartomů (obr. 1). Slizniční léze se vyskytují v dutině ústní a ve střevech (obr. 2). Polypy bývají přítomny téměř u všech pacientů s CS v celém gastrointestinálním traktu. Patří mezi ně hamartomatózní/juvenilní polypy, adenomy, ganglioneuromy a lymfoidní folikuly. Difuzní ezofageální glykogenní akantóza je přítomna u více než 80 % pacientů s CS a v kombinaci s polypózou tlustého střeva může být diagnostická pro CS [10].
Dalším projevem jsou cévní malformace, které mohou postihnout jakýkoli orgán. U pacientů s CS je celoživotní riziko vzniku karcinomu prsu 25–85 %, nádorů štítné žlázy 10–35 %, nádorů endometria 5–28 %, renálního karcinomu 2–34 %, kolorektálního karcinomu 9–20 % a melanomu 0–6 % [6,11,12]. Mezi méně časté patří nádory plic, retinální gliomy, kožní a mozkové nádory. Vzácným tumorem popisovaným v oblasti CNS je dysplastický gangliocytom mozečku (onemocnění Lermit-Duclos).



Specifikace onkogynekologických nálezů asociovaných s CS
Karcinom prsu
Jedná se o nejčastější typ malignity u tohoto syndromu. Naproti tomu byly dosud zaznamenány pouze dva případy karcinomu prsu vyskytujícího se u mužů [6]. U tohoto syndromu jsou často pozorovány fibrotické hyalinizované axilární uzliny s fibrózou a hyalinizací stromatu, a to u 67–89 % pacientek. Pozorovány byly také fibrózní adenomy, duktální hyperplazie a cysty. Zvýšené je riziko multifokálních a oboustranných nádorů [13].


Karcinom endometria
Mutace PTEN genu vede k nadměrnému růstu buněk v děložní sliznici, což zvyšuje riziko vzniku rakovinných změn. PTEN gen také hraje roli v regulaci estrogenní odpovědi a jeho mutace mohou vést k hyperplazii endometria s následným rozvojem atypií až invazivního endometroidního karcinomu těla děložního. Mezi klinické projevy patří zejména nepravidelné krvácení v premenopauze, mohou být přítomny bolesti v oblasti pánve či dyspareunie.
Kazuistika
V naší kazuistice prezentujeme případ 10leté dívky odeslané na gynekologii pro nález patologické rezistence v oblasti levých adnex. V roce 2022 začalo doposud zdravé dívce otékat při fyzické námaze pravé koleno. Občas měla po zátěži potíže s chůzí, nemožnost dřepu či kleku na pravé koleno.
Prvotní vyšetřování probíhalo na ortopedii, kde byl popsán otok pravého kolene, zejména suprapatelárně. Na ultrazvuku (UZ) radiologové popsali synovialitidu pravého kolene. V rámci diferenciální diagnostiky byla vyloučena revmatologická i infekční příčina potíží. V úvahu připadala i juvenilní idiopatická artritida, ale laboratorní nálezy ani klinické projevy nebyly v souladu s touto diagnózou. Současně na očním vyšetření nebyla potvrzena asymptomatická přední uveitida, což je nejčastější revmatické onemocnění v dětském věku a uveitida je jeho nejvýznamnější extraartikulární manifestací.
Z následné magnetické rezonance (MR) dolních končetin dominovala nálezu venolymfatická malformace vycházející z musculus vastus medialis. Po punkci ložiska z biopsie patologové potvrdili mutaci PTEN genu. PTEN gen je tumor supresorový gen lokalizovaný na chromozomu 10q22–23.
Po biopsii následoval odběr krve k vyšetření DNA. V rámci molekulárně genetického vyšetření pomocí Sangerova sekvenování byla u pacientky odhalena mutace PTEN genu v heterozygotní formě, tedy na jedné alele genu. Tím se na molekulární úrovni potvrdil CS. Následně genetické vyšetření v rodině prokázalo, že uvedenou variantu zdědila probandka od svého otce, onemocnění má i sestra dívky.
Vzhledem k cévní malformaci popsané na MR kolene proběhlo hematologické vyšetření k vyloučení vrozených trombofilních rizik. Klinicky byla dívka bez projevů trombózy či abnormálních krvácivých projevů. Z výsledků vyšla izolovaná pozitivita PCG (Pro C global – screeningový test trombofilie) testu, ale bez dalšího patologického nálezu.
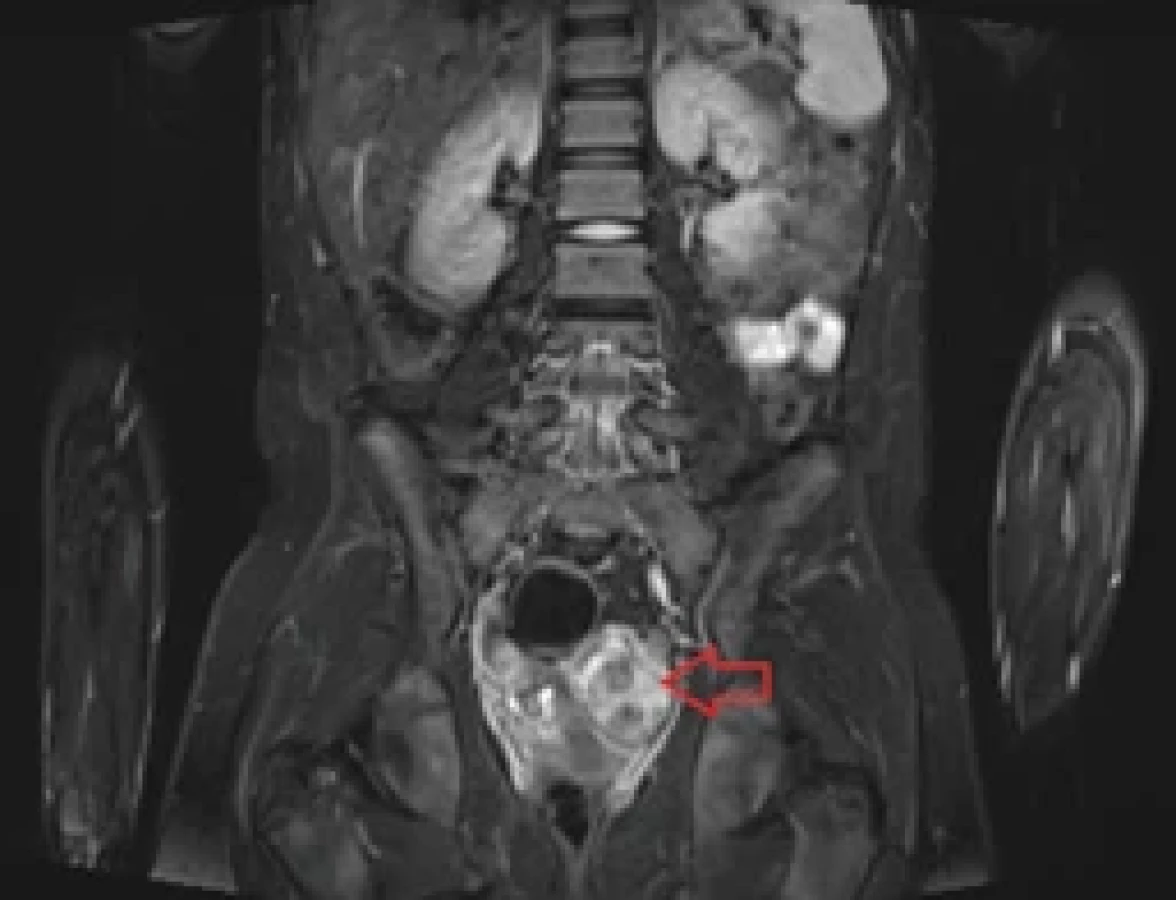
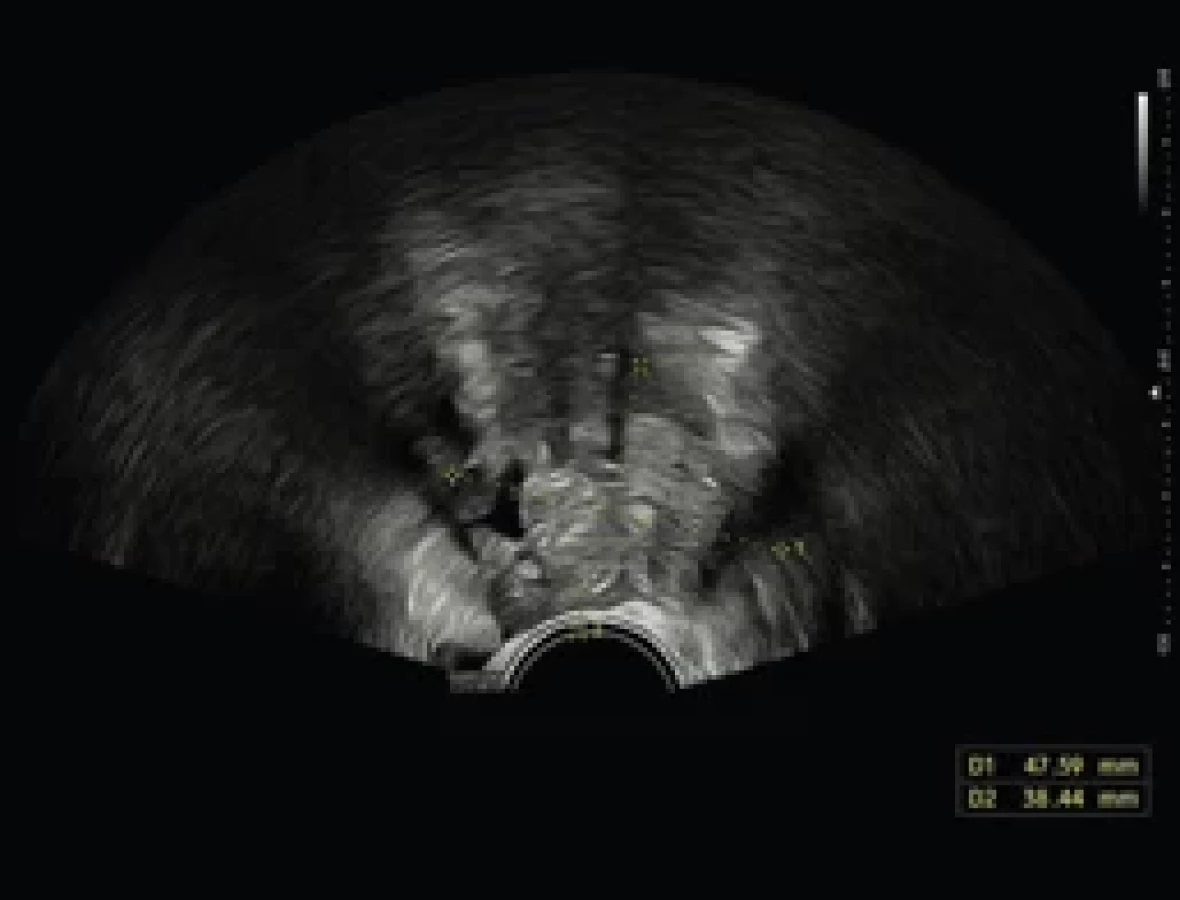
Na základě výsledků genetického vyšetření bylo doplněno celotělové MR. V jeho popisu byla zjištěna malformace v oblasti levého ovaria (obr. 3). Na transvaginálním UZ byla potvrzena heterogenní expanze vel. cca 39 × 38 × 44 mm v oblasti levého ovaria s nehomogenními kalcifikacemi, vrhající akustické stíny, color scale 2, nerovného povrchu, ale mobilní (obr. 4). Dívka neudávala žádné gynekologické potíže, menstruace byla pravidelná, normální intenzity a délky. Na základě stagingu byla indikována diagnostická laparoskopie s laváží. Peroperačně byly zjištěny vícečetné blanité adheze v malé pánvi a expanzivní léze levého ovaria, kde na laterálně lokalizovaný okrsek makroskopicky intaktní ovariální tkáně rozsahu cca 2 × 1 cm s intaktním vejcovodem navazovala plošně přisedlá cystická formace 4 × 3 cm lehce nerovného povrchu s adhezemi k zadní stěně dělohy včetně zadních vazů a k mezorektu. Po postupném rozrušení blanitých adhezí a mobilizaci adnex byla provedena exstirpace cystické léze s resekční linií vedenou makroskopicky intaktní ovariální tkání (obr. 5, 6). Peroperačně z kryobiopsie patologové potvrdili pouze přítomnost benigních cévních malformací bez koincidence s ovariálním tumorem, proto jsme dále již v operačním výkonu nepokračovali. Operační výkon proběhl bez komplikací.
Z definitivního histologického vyšetření resekátu byl prokázán výskyt cévních malformací levého ovaria, což klinicky odpovídá možným a v literatuře uváděným projevům CS (obr. 7). Mikroskopicky byly zastiženy struktury monodermálního teratomu, nezralé struktury ani malignita přítomny nebyly.
Doposud byla dívka bez léčby a od srpna 2024 má na základě mezioborového posouzení a po schválení revizním lékařem nasazenu celkovou léčbu alpelisibem, což je inhibitor proteinové kinázy. Před zahájením terapie ještě byla na kardiologickém vyšetření, které nepotvrdilo žádnou anomálii. Dodržuje režimová opatření a léčbu toleruje dobře.
Vzhledem k UZ nálezu na štítné žláze, kde byly popsány mnohočetné uzlíky, a pro riziko nádorového onemocnění bylo endokrinologem doporučeno její odstranění. V březnu 2025 dívka podstoupila totální tyreoidektomii.
Dívka je v současnosti v péči pracoviště dětské onkologie a dispenzarizace je realizována dle doporučení NCCN pro dětské pacienty s CS. Klinicky se ještě občas vyskytuje bolestivost pravého kolene při fyzické zátěži, ale při zavedené terapii alpelisibem nastala výrazná redukce potíží.
Vzhledem ke genetické zátěži jednoznačně doporučujeme plánované rodičovství s event. využitím metod asistované reprodukce se záměrem preimplantační genetické diagnostiky.


Diskuze
Cowdenův syndrom je vzácné dědičné onemocnění, které je spojeno se zvýšeným rizikem vzniku vícečetných i oboustranných malignit v případě, že se jedná o párový orgán.
Riziko vzniku druhého primárního nádoru je u pacientů s CS až sedminásobně vyšší než v běžné populaci [14]. Tento fakt podtrhuje důležitost pravidelného sledování (tab. 4) a včasného záchytu onemocnění pomocí screeningových metod, které jsou klíčovým nástrojem pro identifikaci nádorů v časných stadiích.
Karcinom prsu byl poprvé jasně rozpoznán jako součást CS v roce 1978 a nyní je považován za nejčastější malignitu u této diagnózy. Screeningová doporučení dle NCCN zahrnují každoroční mammografii a event. magnetickou rezonanci prsu s kontrastem počínající ve věku 30–35 let nebo 5–10 let před nejčasnějším výskytem karcinomu prsu v rodině. Navzdory tomu, že profylaktická mastektomie není doporučena plošně, je vhodné její indikaci zvažovat individuálně [15,16]. Celoživotní riziko karcinomu prsu u žen s CS dosahuje až 85 %, což je srovnatelné s nosiči BRCA mutace [17]. Přitom histopatologické znaky karcinomu prsu nejsou odlišné od běžné populace, což může diagnostiku dále komplikovat.
Mezi další sledované malignity patří karcinom endometria. Riziko vzniku u žen s CS je 5–10 % oproti běžné populaci, kde je 2,6 % [18]. Transvaginální UZ, běžně užívaný u obecné populace, má dle NCCN nízkou specifitu zejména u premenopauzálních žen, proto se jako vhodnější jeví biopsie endometria jednou za 1–2 roky, přestože se jedná o metodu invazivní [18]. Nicméně v současné době je transvaginální UZ provedený zkušeným lékařem základní diagnostickou metodou umožňující detailní zobrazení nálezu v dutině děložní a indikaci bioptického vyšetření při současném posouzení klinických potíží pacientky. Profylaktická hysterektomie rovněž není doporučena jako standardní postup, její indikace je ponechána na individuálním rozhodnutí podle klinické situace [6].
U pacientek s CS je druhým nejčastějším nádorem karcinom štítné žlázy s odhadovaným celoživotním rizikem 38 %, obvykle diagnostikovaným kolem 32. roku života [19]. V naprosté většině případů jde o folikulární či papilární karcinomy. Diagnostika je však komplikována častým výskytem benigních změn, jako jsou solitární uzly či Hashimotova tyreoiditida. Výskyt multinodulární strumy byl důvodem k preventivní tyreoidektomii u námi zmiňované dívky. Screening štítné žlázy se doporučuje již od 7 let věku [16].
Polovina jedinců s CS má adenomatózní nebo hyperplastické kolorektální polypy spojené s časným kolorektálním karcinomem u 13 % pacientů. Proto by se měla provádět rutinní kolonoskopie od 35. roku věku jednou za pět let nebo častěji, pokud má pacient symptomy nebo jsou u něj nalezeny polypy. Screening by však měl začít 5–10 let před věkem výskytu prvních případů kolorektálního karcinomu v rodině.
Riziko renálního karcinomu se odhaduje až na 30 %, a proto se doporučuje ultrasonografie ledvin od 40 let věku v intervalu 1–2 let [16]. U pacientů s CS se také častěji vyskytuje maligní melanom, a to až u 5 % pacientů.
Nespecifické cévní malformace vč. arteriovenózních malformací a hemangiomů se u pacientů s CS vyskytují častěji než v běžné populaci. Míra výskytu cévních malformací v populaci CS se pohybuje od 18 % do 34 % ve srovnání s 5–10 % v běžné populaci [7].
Cévní anomálie jsou v rámci revidovaných klinických kritérií NCCN pro diagnózu PTEN Hamartoma Tumor Syndrom považovány za vedlejší diagnostické kritérium. Uvádí se, že potenciální mechanizmus vzniku cévních lézí u této populace souvisí s úlohou PTEN genu při kontrole angiogeneze spojené s vaskulárním endoteliálním růstovým faktorem. Klinicky se tyto vaskulární léze obvykle projevují jako multifokální, intramuskulární léze s vysokým průtokem nebo jako intrakraniální vývojové abnormality [20]. Napříč modalitami (CT, MR a angiografie) se cévní malformace jeví jako heterogenní léze s abnormální vaskulaturou.
U dívky v popisované kazuistice byly zaznamenány cévní malformace v oblasti pately pravé dolní končetiny a levého ovaria. Navzdory nepřítomnosti některých hlavních kritérií (např. makrocefalie či poruchy autistického spektra) splnila kritéria CS díky přítomnosti dalších významných projevů a potvrzené mutaci PTEN genu. Genetické testování má v diagnostice klíčový význam a mělo by být provedeno na základě indikace klinického genetika a po informovaném souhlasu pacienta.
Přestože je o projevech CS známo mnoho, chybí rozsáhlejší epidemiologická data a přesné odhady výskytu jednotlivých malignit. Většina informací pochází z případových studií a menších souborů publikovaných před zavedením formálních diagnostických kritérií NCCN [20]. Tyto údaje přesto zůstávají zásadním nástrojem pro klinickou praxi a genetické poradenství.


Závěr
Cowdenův syndrom je vzácným genetickým syndromem, jehož diagnostika vychází z klinických projevů, nálezů zobrazovacích metod a následné genetické testace. Dispenzarizace pacientky s CS se opírá o NCCN guidelines. Optimálním postupem je multidisciplinární přístup k dispenzarizaci pacientů s pravidelnými vyšetřeními. Pacienti by měli být informováni o postupech primární i sekundární prevence včetně možností preimplantačního genetického poradenství.

Sources
1. Lloyd KM, Dennis M. Cowden’s disease. A possible new symptom complex with multiple system involvement. Ann Intern Med 1963; 58 (1): 136–142. doi: 10.7326/0003-4819-58-1-136.
2. Magaña M, Landeta-Sa AP, López-Flores Y. Cowden disease: a review. Am J Dermatopathol 2022; 44 (10): 705–717. doi: 10.1097/DAD.00000 00000002234.
3. Ngeow J, Mester J, Rybicki LA et al. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with cowden and cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab 2011; 96 (12): E2063–E2071. doi: 10.1210/jc.2011-1616.
4. Farooq A, Walker LJ, Bowling J et al. Cowden syndrome. Cancer Treat Rev 2010; 36 (8): 577–583. doi: 10.1016/j.ctrv.2010.04.002.
5. Sonagli M, Cagnacci Neto R, da Cruz Formiga MN et al. Cowden syndrome: a single institution case series and literature review. Rev Senol Patol Mamar 2020; 33 (2): 57–60.
6. Takayama T, Muguruma N, Igarashi M et al. Clinical guidelines for diagnosis and management of Cowden syndrome/PTEN hamartoma tumor syndrome in children and adults-secondary publication. J Anus Rectum Colon 2023; 7 (4): 284–300. doi: 10.23922/jarc. 2023-028.
7. Pilarski R. Cowden syndrome: a critical review of the clinical literature. J Genet Couns 2009; 18 (1): 13–27. doi: 10.1007/s10897-008 - 9187-7.
8. Tan MH, Mester J, Peterson C et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet 2011; 88 (1): 42–56. doi: 10.1016/j.ajhg.2010.11.013.
9. Yehia L, Keel E, Eng C. The Clinical spectrum of PTEN mutations. Annu Rev Med 2020; 71 (1): 103–116. doi: 10.1146/annurev-med-0522 18-125823.
10. Plevová P. An update on inherited colon cancer and gastrointestinal polyposis. Klin Onkol 2019; 32 (Suppl 2): 97–108. doi: 10.14735/amko2019S97.
11. Kalin A, Merideth MA, Regier DS et al. Management of reproductive health in Cowden syndrome complicated by endometrial polyps and breast Cancer. Obstet Gynecol 2013; 121 (2 Pt 2 Suppl 1): 461–464.
12. Tischkowitz M, Colas C, Pouwels S et al. Cancer surveillance guideline for individuals with PTEN hamartoma tumour syndrome. Eur J Hum Genet 2020; 28 (10): 1387–1393. doi: 10.1038/s41431-020-0651-7.
13. Puchmajerová A, Vasovcák P, Krepelová A et al. Cowden syndrome. Klin Onkol 2009; 22 (Suppl): S56–S57.
14. Pîrlog LM, Pătrășcanu AA, Militaru MS et al. Insights into clinical disorders in Cowden syndrome: a comprehensive review. Medicina (Kaunas) 2024; 60 (5): 767. doi: 10.3390/medicina 60050757.
15. Frühauf F, Dvořák M, Haaková L et al. Ultrasound staging of endometrial cancer – recommended methodology of examination. Ceska Gynekol 2014; 79 (6): 466–476.
16. Gammon A, Jasperson K, Champine M. Genetic basis of Cowden syndrome and its implications for clinical practice and risk management. Appl Clin Genet 2016; 9 : 83–92. doi: 10.2147/TACG.S41947.
17. Mester J, Eng C. Cowden syndrome: recognizing and managing a not-so-rare hereditary cancer syndrome. J Surg Oncol 2015; 111 (1): 125–130. doi: 10.1002/jso.23735.
18. Dragoo DD, Taher A, Wong VK et al. PTEN hamartoma tumor syndrome/Cowden syndrome: genomics, oncogenesis, and imaging review for associated lesions and malignancy. Cancers (Basel) 2021; 13 (13): 3120. doi: 10.3390/cancers13133120.
19. Tan MH, Mester JL, Ngeow J et al. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 2012; 18 (2): 400–407. doi: 10.1158/1078-0432.CCR-11-2283.
20. Pilarski R. PTEN hamartoma tumor syndrome: a clinical overview. Cancers (Basel) 2019; 11 (6): 844. doi: 10.3390/cancers11060844.
ORCID autorů
J. Pavlacká 0009-0001-3366-6808
M. Felsinger 0000-0002-3826-5675
L. Minář 0000-0001-9088-5428
Doručeno/Submitted: 5. 6. 2025
Přijato/Accepted: 26. 6. 2025
doc. MUDr. Luboš Minář, Ph.D.
Gynekologicko-porodnická klinika
LF MU a FN Brno
Obilní trh 526/11
602 00 Brno
minar.lubos@fnbrno.cz
Labels
Paediatric gynaecology Gynaecology and obstetrics Reproduction medicineArticle was published in
Czech Gynaecology

2025 Issue 5
Most read in this issue
- Vaginální fisting a riziko anogenitálního poranění
- Placentární insuficience a pozdní růstová restrikce ve skupině plodů s adekvátní velikostí pro dané gestační stáří
- Výsledky dotazníku „Endometrióza zdravotního profilu 30“ u žen ve věku 18– 30 v České republice
- Prevalence a hormonální profilování sekundárních amenoreických pacientek dostavujících se na kliniku plodnosti – observační studie