
Development and validation of a HPLC method for quantifica-tion of degradation impurities of salbutamol sulfate with following long-term stability studies in multicomponent cough syrup
Authors:
Ivan Bezruk 1; Anna Materiienko 1; Svitlana Gubar 1; Vera Bunyatyan 2,3; Sergiy M. Kovalenko 2,4; Victoriya Georgiyants 1; Liudas Ivanauskas 5
Authors‘ workplace:
National University of Pharmacy, Department of Pharmaceutical Chemistry
1; Federal State Autonomous Educational Institution of Higher Education I. M. Sechenov First Moscow State Medical University of the Ministry of Healthcare of the Russian Federation, 119991 Moscow, Russia
2; Federal State Budgetary Institution “Scientific Centre for Expert Evaluation of Medicinal Products” of the Ministry of Health of the Russian Federation, 127051 Moscow, Russia
3; V. N. Karazin Kharkiv National University, 61077 Kharkiv, Ukraine
4; Valentynivska Str. , 61100 Kharkiv, Ukraine
4; Lithuanian University of Health Sciences, 44307 Kaunas, Lithuania
5
Published in:
Čes. slov. Farm., 2020; 69, 211-217
Category:
Original article
Overview
Medicines containing both a herbal extract and a synthetic substance are in high demand due to their beneficial effects and synergism. The novel combination of salbutamol sulfate and Hedera helix extracts seems to be prospective in terms of pharmacological activity. But for quality assurance, impurities of the synthetic component have to be determined and quantified. Plant extracts consist of various phytochemical components, therefore, it is more complicated to develop a selective analytical method due to the sample matrix. To prove the safety and efficacy of the dosage form, a new HPLC method for analysis of salbutamol sulfate impurities was developed and validated. The method was used to estimate the safety of the novel syrup by performing long-term stability studies for 24 months. Obtained results indicated the absence in both significant reducing of the main components content and increasing of related substances level. Also, force degradation was carried out to prognosticate the possibility of impurities producing.
Keywords:
long-term stability study – salbutamol sulfate – impurities – multicomponent dosage form – cough syrup – HPLC
Introduction
Respiratory diseases occur in people regardless of age and social status, and coughing remains the most common complaint of patients. An original syrup has been developed based on Hedera helix extract with the addition of salbutamol sulfate1).
Medicines with H. helix leaf extract have advantages in the treatment of acute bronchitis accompanied by cough and sputum due to triterpene saponins, which show a mucolytic, mucokinetic, and antispasmodic action2, 3). It is a well-known fact that for better expectoration of sputum it is necessary to stimulate the enlargement of the bronchi. In this case, in addition to multicomponent medicine bronchodilator substances, such as salbutamol sulfate, can be quite useful in the treatment of bronchopulmonary diseases. Previously, the quality of the novel syrup was confirmed by analysing of the H. helix main component1).
It is worth mentioning that different components, intermediates, and reagents are used during the synthesis of active pharmaceutical ingredients (API). Thus, identification, quantification, and control of impurities in the synthetic substance and its dosage forms play a critical role in providing patients with safe medicines4, 5). The method for analysis of salbutamol sulfate is reported in EP6). Some other literature sources propose the HPLC for quantification of related substances7–10) as well as enantiomeric separation11) of salbutamol sulfate.
Also, the LC-MS12) and GC-MS13) might be used. However, none of the above-mentioned methods describe the analysis of salbutamol sulfate impurities in combination with a plant extract which can be challenging due to the content of different phytochemicals. Moreover, no publication has established stability in studying storage dosage forms with salbutamol sulfate for long terms. This study aimed to estimate the safety of the novel combination and to determine the main impurities profile for salbutamol sulfate.
Experimental part
Instrument
Liquid chromatography separation was performed using a Shimadzu Nexera X2 LC-30AD HPLC system (Shimadzu, Japan) composed of a quaternary pump, an on-line degasser, a column temperature controller, a SIL-30AC autosampler (Shimadzu, Japan); a CTO-20AC thermostat (Shimadzu, Japan) as well as a SPD-M20A diode array detector (DAD). Other instruments such as an Ultrasonic Cleaner Set for ultra-sonication using (Wise Clean WUC-A06H, Witeg Labortechnik GmbH, Germany), a Libra UniBloc AUW120D (Shimadzu Analytical Scale, Japan); a рН-meter – a Knick Electronic Battery-Operated pH Meter 911 PH (Portamess, Germany), and class A analytical volumetric flasks that meet the requirements of the SPhU were used in the investigation.
Chemicals and reagents
HPLC grade methanol and acetonitrile, analytical grade hydrogen peroxide, hydrochloric acid, sodium hydroxide, acetic acid, and triethylamine (TEA) were from Merck (Darmstadt, Germany). Salts of ammonium acetate, sodium dihydrogen phosphate, and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich GmbH (Steinheim, Germany). Salbutamol sulfate EPCRS, impurity-G EPCRS, impurity-D EPCRS, impurity-F EPCRS were purchased from EP commission. Purified deionized water was produced with a Millipore (Burlington, MA., USA) water purification system.
Syrup compositions
All series of syrups were prepared by Vishpha (Zhytomyr, Ukraine) as follows (amount of substance in dosage form are specified in percents): extract H. helix leaf (0.45%) and salbutamol sulfate (0.048%) as active pharmaceutical ingredients and sorbitol (35%), citric acid (0.045%), potassium sorbate (0.2%), natural gum (0.22%), and purified water as excipients.
Chromatographic conditions
Separation of compounds was performed with an ACE C18 column (150*4.6 mm, particle size 5 µm). The binary solvent system was used: 0.05% TEA solution adjusted to 5.5 pH with acetic acid as the mobile phase A and a mixture of methanol: acetonitrile 50 : 50 (v/v) as the mobile phase B with the flow rate of 1.0 ml/min. The following linear gradient elution was used: 95% A/5% B – 0 min, 95% A/5% B – 5 min, 10% A/90% B – 30 min, 10% A/90% B – 32 min, 95% A/5% B – 34 min. The detection of components was done at 277 nm. The column oven temperature was set at 35 °C.
The preparation of the solution for system suitability
The final concentrations of components were as follows: for salbutamol sulfate 400 µg/ml, potassium sorbate 2 mg/ml, 1.8 µg/ml for impurities, D, F, G in water.
Test solution
15.0 ml of syrup were placed into a 20.0 ml volumetric flask and made up to mark with water. All solutions were filtered through a 0.45 µm membrane filter.
Stability studies
Syrup stability was tested according to ICH guidelines, at the temperature 25 ± 2 °C and humidity 60 ± 5% maintained by a thermostat. Three different batches of syrup were stored at the mentioned conditions in amber glass bottles. The impurity profile was measured directly following preparation and after 3, 6, 9, 12, 15, 18, 21, 24 months.
Degradation studies
The concentration of salbutamol sulfate for all stress studies was the same as in test solution. Alkaline hydrolysis was performed with adding of 2 mL of 0.1 М NaOH at ambient temperature for 3 hours. The effect of acidic decomposition was tested with 2 mL of 0.1 М HCl at the ambient temperature for 3 hours. Oxidative study was performed with 2 mL of 3 % hydrogen peroxide at the ambient temperature for 3 hours. Also, the temperature degradation was used, the sample was heated at 100 °C for 3 hours. Samples were withdraw at appropriate time and analysed with the proposed HPLC method.
Results
Method development
The possible impurities of salbutamol sulfate are similar to the main component14). To achieve a suitable resolution among all components in the novel composition, different conditions were tested. In particular, the influence of different stationary phases sorbents (C8 and C18) as well as their particle size (3 µm and 5 µm) were studied. The C18 stationary phase with particle size 5 µm was recognized as the most suitable for the separation of components in the studied combination.
Moreover, the influence of the mobile phase buffer salt and additives were investigated. Two different salts in various concentrations were tested: ammonium acetate (5 mM – 25 mM) and sodium dihydrogen phosphate (5 mM – 50 mM) with different pH values 3.0–6.0. However, the resolution of some impurities with other components of combination remained unsatisfactory. The salts were replaced with additives such as triethylamine (TEA) and trifluoroacetic acid (TFA). Water solution with TEA adjusted to pH 5.5 showed the most suitable separation among components.
Methanol and acetonitrile were chosen as components of the mobile phase B. Pure methanol gave a poor baseline with a baseline drift and peak shapes were not acceptable. Hence acetonitrile was used to correct it. Ration 50/50 (v/v) was the most suitable.
Method validation
A new HPLC method for analysis of salbutamol impurities has been developed and validated according to State Pharmacopeia of Ukraine15) and ICH16). There are many developed methods for the determination of salbutamol sulfate impurities. However, their applying for analysis of the cough syrup was not so successful, since a poor separation between the syrup matrix and salbutamol impurities has been observed.
System suitability test
The observed RSD values at a 1 % level of analyte concentration were within suitable values (≤ 2%). The resolution, number of the theoretical plate, and tailing factor for all main components were determined. All values were within suitable limits.
Specificity
Specificity studied has shown that no peak either from placebo of the syrup matrix or from forced degradation studies was coeluted at the retention time of salbutamol sulfate and its impurities. The identification of components was done by comparing the retention times of peaks from the test solution with the peaks of the standard solution.
Limit of detection
Investigation of the sensitivity of the method has been carried out by analysis of a prepared solution with a concentration of salbutamol sulfate (36 ng/mL) which was 0.01% from its amount in the test solution. Obtained ratio of signal/noise for the main peak was greater than 3/1 hence the method was approved as sensitive.
Robustness
Examination of robustness has been performed with a changing temperature oven (30–40 °C), flow rate (0.9 ml/min to 1.1 ml/min), and pH of mobile phase A (5.0–6.0) (Table 1, Fig. 1). Obtained results showed that the main effect on the system was exerted by pH of the Mobile phase, thus at high values of pH, no separation between peaks of salbutamol sulfate and excipients was observed.


Stability studies
Long-term stability studies were performed using the proposed method. The results obtained from stability studying are shown in Table 2, Fig. 2.


According to the requirements for impurities level, there are such limits in an amount: for both impurity D and G not greater than 0.5% (from the amount of salbutamol sulfate in the dosage form), for impurity F not greater than 0.3 %, unspecified impurity not greater than 0.1%, and the sum of impurities not greater than 1.5 %.
The obtained results indicate that syrup contained only impurity F and some unspecified impurities, moreover their concentrations change insignificantly. Other impurities were not produced under storage conditions. Thus, the novel syrup was found stable for 24 months of storage, since the amount of each impurity was lesser than the allowed level.
Forced degradation study
Forced degradation was performed on salbutamol sulfate substance to prove the specificity of the method and to determine the impurities profile. Four different degradations were attempted, such as alkaline, acidic, temperature, and peroxidic. The stability of composition was carried out by preparing the sample solution as mentioned above and injected at regular parameters. From the forced degradation study it was observed and confirmed that no other formulation components and potential degradation product and impurities had interfered in the determination. The results of degradation are shown in Table 3, Fig. 3.

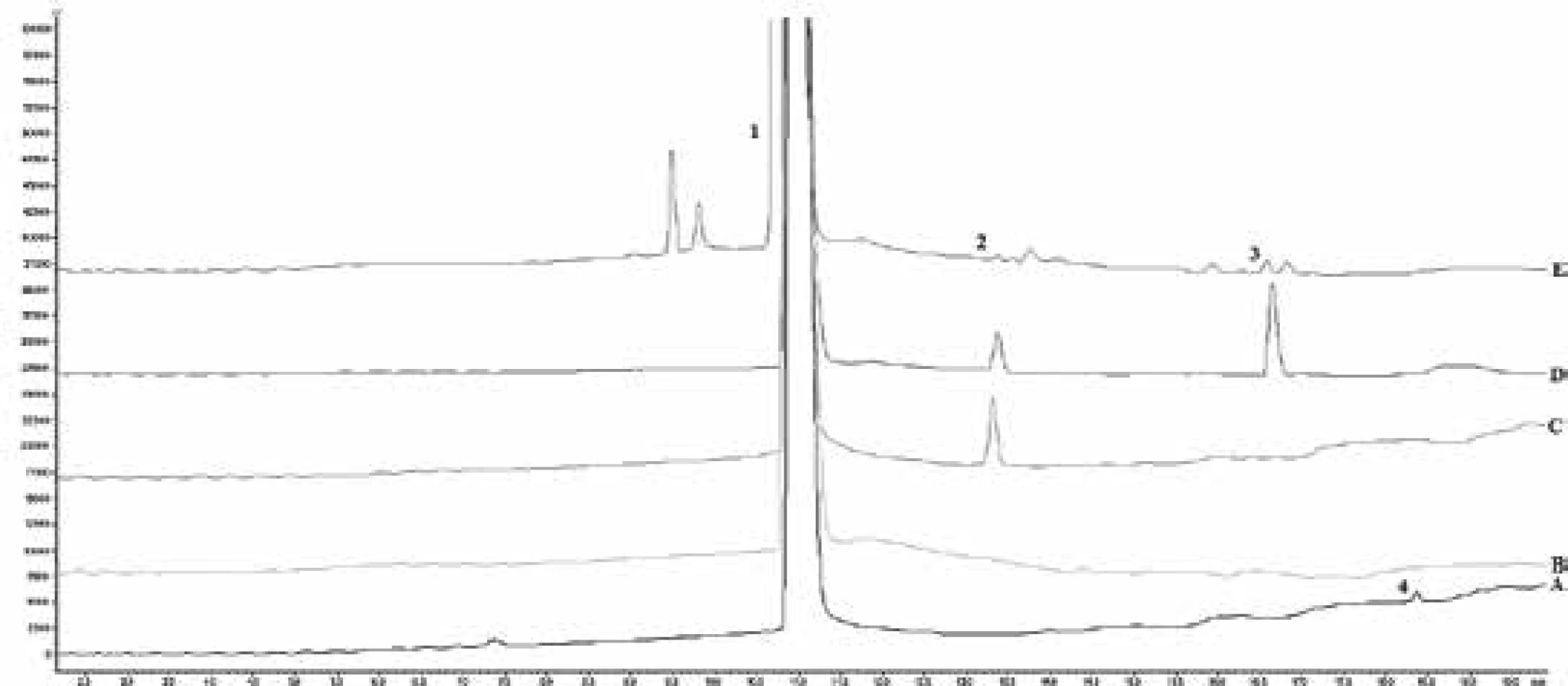
As can be seen, salbutamol sulfate was not stable to the effect of temperature, alkaline hydrolysis, and oxidation. Peroxide influence had the greatest effect on producing different unspecified impurities. On the other hand, the highest level of impurity D and impurity G was after the alkaline effect on the solution.
Conclusions
A new HPLC method for the determination of salbutamol sulfate impurities was developed and validated. The developed method was used in long-term stability studies of a novel cough syrup. Obtained results showed that salbutamol sulfate is stable in syrup during all terms of storage (24 months), however, it degrades under temperature, alkaline, and oxidation actions. The main observed related substances of salbutamol sulfate were impurity D, impurity G, and impurity F. On the other hand, only impurity F was present in the syrup, while others were detected only after degradation.
Conflict of interest: none.
Received July 29, 2020 / Accepted October 12, 2020
Ivan Bezruk – PhD student (∗) • A. Materiienko • S. Gubar • V. Georgiyants
National University of Pharmacy
Department of Pharmaceutical Chemistry
Valentynivska Str. 4, 61100 Kharkiv, Ukraine
e-mail: vania.bezruk@gmail.com
V. Bunyatyan1,2, S. M. Kovalenko1,3
1Federal State Autonomous Educational Institution of Higher Education I. M. Sechenov First Moscow State Medical University of the Ministry of Healthcare of the Russian Federation, 119991 Moscow, Russia
2Federal State Budgetary Institution “Scientific Centre for Expert Evaluation of Medicinal Products” of the Ministry of Health of the Russian Federation, 127051 Moscow, Russia
3V. N. Karazin Kharkiv National University, 61077 Kharkiv, Ukraine
L. Ivanauskas
Lithuanian University of Health Sciences, 44307 Kaunas, Lithuania
Sources
1. Bezruk I., Kotvitska A., Korzh I., Materiienko A., Gubar S., Budanova L., Ivanauskas L., Vyshnevsky I., Georgiyants V. Combined Approach to the Choice of Chromatographic Methods for Routine Determination of Hederacoside C in Ivy Leaf Extracts, Capsules, and Syrup. Sci. Pharm. 2020; 88(2), 24.
2. Greunke C., Hage-Hülsmann A., Sorkalla T. A systematic study on the influence of the main ingredients of ivy leaves dry extract on the β2-adrenergic responsiveness of human airway smooth muscle cells. Pulmonary Pharmacology & Therapeutics. 2015; 31, 92–98.
3. Hofmann D., Hecker M. Efficacy of dry extract of ivy leaves in children with bronchial asthma – a review of randomized controlled trials. Phytomedicine 2003; 10, 213–220.
4. Mauryaand C. P., Lokhande M. V. Impurity Profile and Validation of Pharmaceutical (API) Bulk Drug. World Journal of Pharmaceutical Research 2017; 6(11), 878–887.
5. Churiand S. K., Lokhande M. V. Identification and Impurity Profiling of Process Related Impurities In DTPEE. European Journal of Biomedical and Pharmaceutical Sciences. 2017; 4(9), 617–623.
6. European Pharmacopoeia 10th ed. 2019. Strasbourg.
7. Erram S. V., Fanska C. B., Asif M. Determination of albuterol sulfate and its related substances in albuterol sulfate inhalation solution, 0.5% by RP-HPLC. J. Pharm. Biomed. Anal. 2006; 40, 864–874.
8. Malkki L., Tammilehto S. Optimization of the separation of salbutamol and its decomposition products by liquid chromatography with diode-array detection J. Pharm. Biomed. Anal. 1993; 11, 79–84.
9. Beaulieu N., Cyr T. D., Lovering E. G. Liquid chromatographic methods for the determination of albuterol (salbutamol), albuterol sulfate, and related compounds in drug raw materials, tablets, and inhalers. J. Pharm. Biomed. Anal. 1990; 8, 583–589.
10. Ong H., Adam A., Perreault S., Marleau S., Bellemare M., Du Souich P., Beaulieu N. Analysis of albuterol in human plasma based on immunoaffinity chromatographic clean-up combined with high-performance liquid chromatography with fluorimetric detection. J. Chromatogr. 1989; 497, 213–221.
11. Halabi A., Ferrayoli C., Palacio M., Dabbene V., Palacios S. Validation of a chiral HPLC assay for (R)-salbutamol sulfate J. Pharm. Biomed. Anal. 2004; 34, 45–51.
12. Zhang J., Xu Y., Di X., Wu M. Quantitation of salbutamol in human urine by liquid chromatography–electrospray ionization mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2006; 831, 328–332.
13. Weisberger M., Patrick J. E., Powell M. L. Quantitative analysis of albuterol in human plasma by combined gas chromatography chemical ionization mass spectrometry Biomed. Mass Spectrom. 1983; 10, 556–558.
14. Kasawar G. B., Farooqui M. Development, and validation of a stability-indicating RP-HPLC method for the simultaneous determination of related substances of albuterol sulfate and ipratropium bromide in nasal solution. Journal of Pharmaceutical and Biomedical Analysis 2010; 52, 19–29.
15. State Pharmacopoeia of Ukraine 2nd ed. 1st supplement. Kharkiv: State Enterprise “Scientific-and-expert Pharmacopeial Centre” 2015; 1128 pp.
16. ICH harmonized tripartite guideline Q2 (R1). Validation of analytical procedures: text and methodology Q2 (R1), in Proceedings of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 2005.
Labels
Pharmacy Clinical pharmacologyArticle was published in
Czech and Slovak Pharmacy

2020 Issue 5-6
Most read in this issue
- Meropenem serum concentrations in intensive care patients: a retrospective analysis
- Automated preparation of radiopharmaceuticals as a tool of radiation protection optimisation of staff
- K životnímu jubileu prof. RNDr. Jozefa Csölleiho, CSc.
- Development and validation of a HPLC method for quantifica-tion of degradation impurities of salbutamol sulfate with following long-term stability studies in multicomponent cough syrup