
Transplantace jater pro deficit alfa-1-antitrypsinu
Liver transplantation in alpha-1-antitrypsin defi ciency
Alpha-1-antitrypsin deficiency (AATD) is an autosomal codominant genetic condition which manifests with lung emphysema at young age and with liver disease. A point mutation in the SERPINA1 gene, which encodes a1-antitrypsin (AAT), causes a defective secondary structure of the protein, which subsequently accumulates in the endoplasmic reticulum of hepatocytes and is not transported into the blood and body fluids. Deficiency of AAT, which inhibits neutrophil elastase in the lung, thus allows proteolytic damage to the connective tissue of the lung, leading to the development of emphysema. Liver disease is caused by the accumulation of the mutant AAT in liver cells, contributing to proteotoxic liver injury that can lead to liver cirrhosis. Liver transplantation (LT) is the only curative method; after LT, the recipient has the phenotype of the donor organ and normal serum AAT concentrations. The indication for LT should be considered in all patients with AATD and end-stage liver disease. The patients should be referred to the transplant centre when severe complications of liver cirrhosis, such as variceal bleeding, ascites, encephalopathy, and hepatorenal syndrome. In patients with hepatocellular carcinoma, LT represents an optimal treatment modality in the case of an early, unresectable tumour. The timing of LT is crucial; LT should be performed before the patient develops life-threatening complications of end-stage liver disease. LT should be considered in patients of Child-Pugh classification B and C and MELD score of 15 points or higher. Survival of patients after LT for AADT is excellent.
Keywords:
alpha-1-antitrypsin – alpha-1-antitrypsin deficiency – liver cirrhosis – end-stage liver disease – hepatocellular carcinoma – live transplantation
Autoři:
S. Fraňková 1
![]() ; M. Holinka 1,2
; M. Holinka 1,2
![]() ; J. Šperl 1,3
; J. Šperl 1,3
![]()
Působiště autorů:
Klinika hepatogastroenterologie, Transplantcentrum, Institut klinické a experimentální medicíny, Praha 2 3. LF UK, Praha 3 1. LF UK, Praha
1
Vyšlo v časopise:
Gastroent Hepatol 2024; 78(2): 129-135
Kategorie:
Hepatologie
doi:
https://doi.org/10.48095/ccgh2024129
Souhrn
Deficit a1-antitrypsinu (AATD) je autozomálně kodominantní onemocnění projevující se rozvojem emfyzému plic v mladém věku a onemocněním jater. Bodová mutace genu SERPINA1, který a1-antitrypsin (AAT) kóduje, způsobuje defektní sekundární strukturu proteinu, který se následně hromadí v endoplazmatickém retikulu hepatocytů a není transportován do krve a tělesných tekutin. Nedostatek AAT, který blokuje neutrofilní elastázu v plicích, tak umožní proteolytické poškození pojivové tkáně plic, což vede k rozvoji emfyzému plic. Jaterní onemocnění je způsobeno hromaděním mutovaného AAT v jaterních buňkách, což vede k proteotoxickému jaternímu poškození, které může vést až k jaterní cirhóze. Transplantace jater (LT) představuje jedinou kurativní metodu, po LT má příjemce jaterního štěpu fenotyp dárce orgánu a normální sérové koncentrace AAT. Indikace k LT má být zvážena u všech pacientů s AATD a konečným stadiem selhání jater. Tito nemocní mají být odesláni do transplantačního centra v okamžiku vzniku závažných komplikací, jako jsou variceální krvácení, ascites, encefalopatie, hepatorenální syndrom. U pacientů s hepatocelulárním karcinomem je LT optimální léčebnou metodou v případě časného, neresekabilního tumoru. Načasování LT je zcela zásadní, LT má být provedena dříve, než pacient vyvine život ohrožující komplikace jaterního onemocnění. LT má být zvažována u pacientů Child- -Pughovy klasifikace B a C a při MELD (Model of End-Stage Liver Disease) skóre 15 a vyšším. Přežití nemocných po LT je v současné době excelentní.
Klíčová slova:
Alpha-1-antitrypsin – deficit alpha-1-antitrypsinu – jaterní cirhóza – jaterní selhání – hepatocelulární karcinom – transplantace jater
Úvod
Deficit a1-antitrypsinu (AATD) je autozomálně kodominantní onemocnění projevující se rozvojem emfyzému plic v mladém věku a onemocněním jater [1]. Bodová mutace genu SERPINA1, který a1-antitrypsin (AAT) kóduje, způsobuje defektní sekundární strukturu proteinu, který se následně hromadí v endoplazmatickém retikulu (ER) hepatocytů a není transportován do krve a tělesných tekutin. V ER dochází k polymerizaci proteinu. Nedostatek AAT („loss of function“) tak umožní proteolytické poškození pojivové tkáně plic, což vede k rozvoji emfyzému. Naopak jaterní onemocnění je způsobeno hromaděním mutovaného AAT v jaterních buňkách („gain of function“), což způsobuje proteotoxické jaterní poškození, které může vést k rozvoji jaterní cirhózy a hepatocelulárního karcinomu (HCC). AATD je nejčastějším genetickým jaterním onemocněním u dětí. V dospělosti způsobuje klinicky významné onemocnění jater u jedinců s mutací v homozygotní konstituci ve třetí dekádě života – je tedy zřejmé, že významnou roli v rozvoji jaterního onemocnění hrají i další genetické faktory a faktory prostředí. Transplantace jater (LT) je dosud jedinou účinnou léčbou u jedinců s jaterním selháním při AATD, nicméně v blízké budoucnosti můžeme očekávat nové možnosti léčby pomocí genové terapie.
![Sérové koncentrace u klinicky nejvýznamnějších genotypů AAT, upraveno podle [3].](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image/6dc3a46afae2a29530b30ad58d1f4be9.png)
Patogeneze
Příčinou AATD jsou mutace v genu SERPINA1 kódujícím AAT. AAT je tvořen v největší míře v hepatocytech a je vylučován do krevního oběhu, aby v plicích plnil ochrannou funkci před proteolytickou degradací elastázou neutrofilů. Základním znakem AATD v jaterní biopsii je přítomnost granul pozitivních v barvení periodic acid-Schiff, rezistentních k diastáze (tzv. PAS-D pozitivní granula). Jedná se o polymerizovaný, abnormálně složený protein, který zůstává v hepatocytu, jenž není schopen opustit. Akumulace AAT v ER je patogenetickým mechanizmem jaterního onemocnění: nadbytek polymerizovaného proteinu vysoce přesahuje kapacitu degradace a makroautofagie ER, dochází k polymerizaci a akumulaci AAT, což vede k proteotoxickému stresu, poruše funkce ER, k zániku hepatocytů, zánětu, fibróze a cirhóze [2].
Proteolytické poškození plic je způsobeno nízkou koncentrací AAT, která vede k nedostatečné inhibici neutrofilní elastázy v plicích. Klinicky se AATD manifestuje jako časný panlobulární plicní emfyzém a/anebo chronická obstrukční plicní nemoc (CHOPN). Postižení plic a jater představují zásadní příčiny mortality při AATD.
Koncentrace AAT v séru je závislá na genotypu jedince (tab. 1).
Wild type („zdravé“) alely AAT zvané M reprezentují kteroukoli z pěti polymorfních alel M1A, M1V, M2–M4, s identickou migrací proteinu při izoelektrické fokusaci. Genotyp Pi*MM v homozygotní konstituci je přítomen přibližně u 94,5 % zdravé populace ČR [4].
Variantní (patogenní) alely můžeme rozdělit na tři skupiny.
Prvním typem jsou tzv. storing (střádavé) alely, nejčastější je variantní alela Z (p.Glu366Lys, rs28929474 c.1096G>A), a dvě vzácné varianty Mmalton (p.Phe76del, rs775982338 c.226_228delTTC) a Siiyama (p.Ser77Phe, rs55819880 c.230C>T) [5,6]. Tvořené proteiny podléhají jak částečné intracelulární degradaci (70 %), tak sekreci (15 %) a tvorbě polymerů (15 %). Pouze malá část již vytvořených polymerů je degradována nebo vyloučena mimo buňku. Zbytek zůstává v ER a tato depozita jsou detekovatelná jako PAS-D pozitivní inkluze v histologickém obraze [7]. Homozygotní mutace, tzv. Pi*ZZ genotyp, je odpovědná za většinu případů AATD a vede k jaternímu poškození jak u dětí, tak i u dospělých.
Druhým typem jsou tzv. nulové alely charakterizované absencí tvorby proteinu nebo syntézou nepolymerizujícího zkráceného proteinu.
S alela (p.Glu288Val, rs17580 c.863A>T) reprezentuje třetí typ variantní alely charakterizovaný syntézou dysfunkčního proteinu, který však podléhá kompletní intracelulární degradaci.
Epidemiologie
Incidence AATD je 1/3 000 narozených dětí ve většině studovaných populací [8]. Většina studií provedených na pacientech s jaterním nebo plicním onemocněním byla ovlivněna zařazením pacientů, kteří byli sledováni ve specializovaných centrech, plicních nebo hepatologických. Švédská studie ze 70. let 20. stol. prováděla screening novorozenců na AATD: vyšetřeno bylo více než 200 tis. novorozenců a bylo nalezeno 127 AAT Pi*ZZ homozygotů, kteří byli dále sledování téměř 30 let [9,10]. Čtrnáct (11 %) z těchto pacientů mělo protrahovanou novorozeneckou žloutenku a pouze devět jedinců (7 %) vyvinulo ve sledovaném období klinicky významné onemocnění jater. Ve studii však nebyly prováděny biopsie jater a klinicky významné jaterní onemocnění se projeví obvykle ve věku 50–65 let. Onemocnění plic postihne významnou část Pi*ZZ homozygotů, nicméně konkrétní závěry lze rovněž učinit až v okamžiku, kdy sledovaná kohorta pacientů dosáhne vrcholu věku pro rozvoj emfyzému, tedy 40–60 let [11].
Klinická manifestace onemocnění
Většina dětí s AATD je klinicky zdravá. Jaterní onemocnění se může projevit ve věku 4–8 týdnů protrahovanou žloutenkou. Laboratorně nacházíme zvýšené hodnoty konjugovaného bilirubinu a jaterních aminotransferáz. Játra mohou být zvětšena, mohou být rovněž přítomny krvácivé projevy, ale málokdy je přítomna závažná syntetická jaterní dysfunkce. Na AATD je proto nutno pomýšlet v rámci diferenciální diagnózy neonatálních hepatitid [12–14].
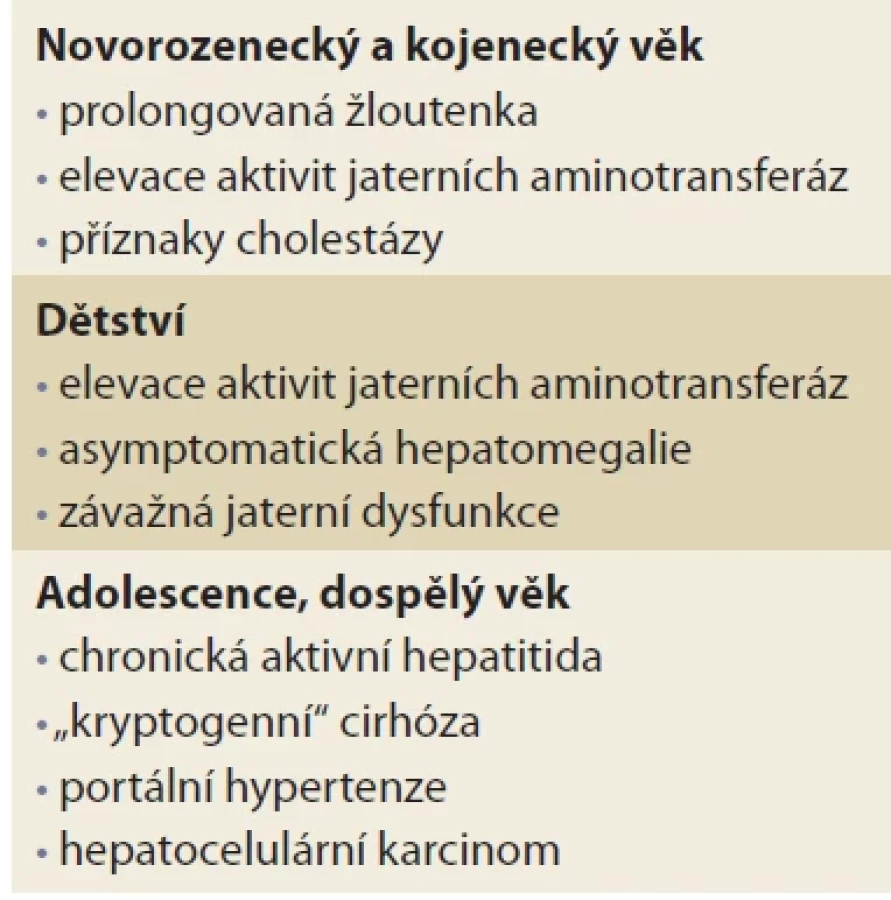
AATD může být diagnostikován u starších dětí a může se projevit hepatomegalií, náhodným nálezem zvýšených aktivit jaterních enzymů nebo ikterem při interkurentní infekci. Onemocnění se však může poprvé projevit komplikacemi portální hypertenze: splenomegalií, hypersplenizmem, krvácením z varixů jícnu nebo žaludku, ascitem nebo jaterní encefalopatií. Vyšetření AAT by mělo být proto nedílnou součástí diferenciální diagnostiky všech dospělých pacientů s chronickým onemocněním jater, kryptogenní cirhózou nebo nově zjištěným HCC [15,16]. Riziko rozvoje závažné jaterní fibrózy a cirhózy je u Pi*ZZ homozygotů 20krát vyšší ve srovnání se zdravými jedinci. K progresi jaterního onemocnění u AATD přispívá mužské pohlaví, přítomnost steatózy, diabetes mellitus, abúzus alkoholu a vyšší věk. Riziko cirhózy je vyšší u nekuřáků, kteří se díky pomalejší progresi plicního onemocnění dožijí vyššího věku [16,17]. Klinické projevy onemocnění v závislosti na věku jsou přehledně uvedeny v tab. 2.
Heterozygotní nosičství Pi*MZ genotypu SERPINA1 představuje spíše rizikový faktor než jasnou predispozici k jaternímu onemocnění, je třeba ještě dalšího faktoru, který jaterní onemocnění způsobí [16]. Nosiči Pi*MZ genotypu mají 1,7krát vyšší riziko úmrtí v souvislosti s jaterním onemocněním ve srovnání se zdravými jedinci. Nosičství Pi*MZ genotypu zvyšuje riziko rozvoje jaterní cirhózy u pacientů s alkoholickou chorobou jater, nealkoholickou steatohepatitidou a cystickou fibrózou [18].
Plicní postižení u nemocných s AATD se obvykle projeví ve třetí až čtvrté dekádě dušností a jinými symptomy, jako jsou kašel, zahlenění, sípaní a slabost [1]. Mnoho pacientů s AATD nemá symptomy žádné a onemocnění je odhaleno v rámci screeningu rodinných příslušníků. K rozvoji symptomů dochází v mladším věku než u pacientů s CHOPN a často u nich chybí kuřácká anamnéza. U symptomatických pacientů jsou patrny známky obstrukční ventilační poruchy s poklesem FEV1 (usilovně vydechnutý objem za 1 vteřinu), se vzestupem reziduálního objemu a je též přítomna porucha difuze.
Diagnóza AATD
Diagnóza deficitu AATD by měla být zvažována u všech pacientů se zvýšenou aktivitou jaterních aminotransferáz, konjugovaného bilirubinu, u pacientů s asymptomatickou hepatomegalií, známkami portální hypertenze nebo cholestázy, u nemocných s prodlouženým protrombinovým časem. U dospělých je třeba myslet na AATD u jedinců s chronickou hepatitidou, jejíž původ nebyl spolehlivě objasněn, u kryptogenních cirhóz a pacientů s HCC.
Základním vyšetřením je stanovení sérové koncentrace AAT. Ta je obvykle stanovena za pomoci přidání protilátky, která se naváže na AAT s následným využitím turbidimetrie. Na AATD můžeme mít rovněž podezření při nálezu snížení koncentrace proteinů alfa-1 frakce při elektroforéze sérových bílkovin.
V minulosti se ke stanovení genotypu využívala izoelektrická fokusace: na pohyblivosti mutovaného proteinu při izoelektrické fokusaci je proto založena historická, ale stále nejčastěji používaná nomenklatura s označováním variant Pi*M, Pi*S, Pi*Z (protein M, produkt divoké, tedy zdravé alely se pohybuje ve středu elektrického pole – middle).
Diagnostika genetická, dnes široce dostupná, využívá genotypizace pomocí RT-PCR nebo sekvenace genu SERPINA1 a diagnózu AATD definitivně potvrdí.
Histopatologická diagnostika AATD je možná ze vzorku jaterní tkáně získané jaterní biopsií či při vyšetření explantátu při LT. Polymery mutovaného AAT prokazujeme v hepatocytech jako PAS-D pozitivní agregáty, typicky v periseptální lokalizaci. Definitivně diagnózu potvrdí imunohistochemické vyšetření pomocí polyklonální králičí protilátky (obr. 1).
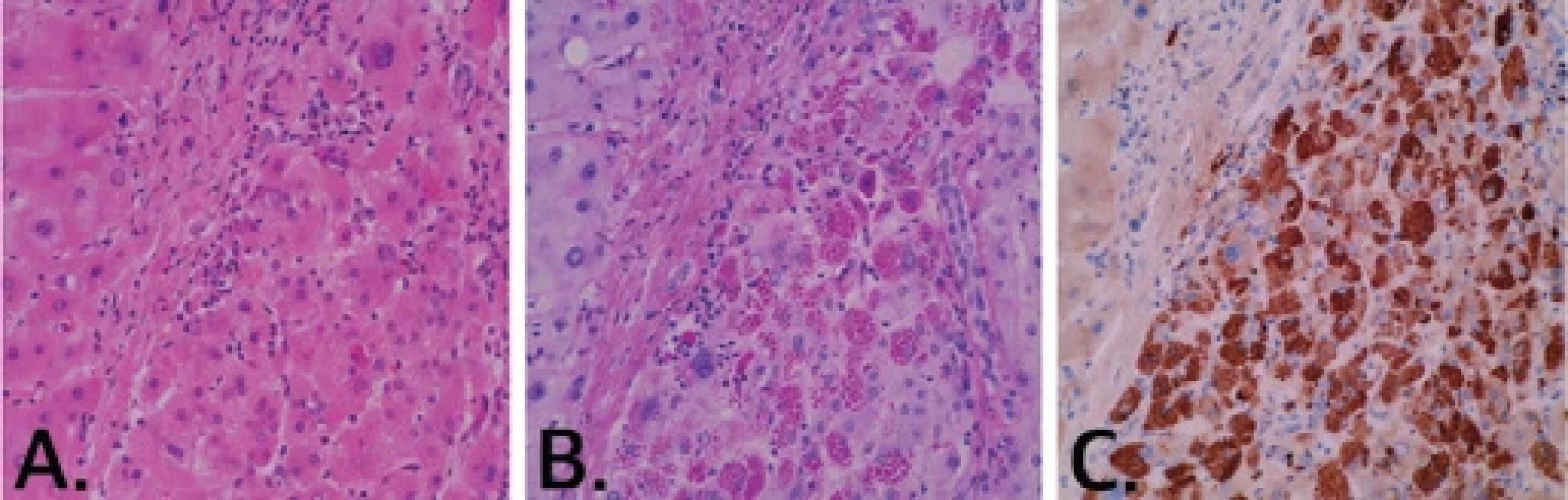
A) Hematoxylin-eozin (zvětšeno 400×); B) barvení PAS-D (zvětšeno 400×); C) imunohistochemické vyšetření pomocí polyklonální králičí protilátky (zvětšeno 400×).
A) Hematoxylin-eosin staining (magnifi cation 400×); B) PAS-D staining (magnifi cation 400×); C) immunohistochemical staining with the polyclonal rabbit antibody (magnifi cation 400×).
Léčba plicního onemocnění
Purifikovaný lidský AAT je izolován z krevní plazmy a lze jej využít jako augmentační léčbu u emfyzému plic, který je způsoben deficitem funkčního cirkulujícího AAT [19]. Je podáván intravenózně. V České republice je léčba hrazena u pacientů s chronickou CHOPN stadia III a IV, s geneticky prokázaným deficitem AAT, sníženou sérovou koncentrací AAT < 0,5–0,8 g/l a hodnotou FEV1 < 60 % náležitých hodnot. Podmínkou také je, že pacienti nesmí kouřit. U pacientů v terminálním stadiu respirační insuficience je nutno zvažovat transplantaci plic.
Léčba jaterního onemocnění
Pacienti s AATD mají dodržovat režimová opatření, která mohou ovlivnit další faktory přispívající k progresi jaterního onemocnění. Zásadní je abstinence od alkoholu, udržování ideální tělesné hmotnosti a těsná kompenzace diabetu mellitu, pokud je přítomen [16].
Genová terapie je rychle se vyvíjející oblast léčby vrozených onemocnění, která může být využita i v terapii AATD. V současnosti se zdá slibné využité interferující RNA – lék fazirsiran (Arrowhead Pharmaceuticals/Takeda Pharmaceuticals, USA) je v současnosti ve fázi III klinického testování. Jedná se o subkutánně aplikované injekce. Fazirsiran způsobuje degradaci AAT a Z-AAT mRNA, což vede ke snížení tvorby AAT a Z-AAT v hepatocytech [20].
LT je dosud jedinou účinnou léčbou u jedinců s jaterním selháním při AATD.
Transplantace jater pro AATD: počáteční zkušenosti
První úspěšná LT pro AATD byla provedena v roce 1973 u 16leté dívky s jaterní cirhózou. Dříve popsané dva případy z počátku 70. let skončily úmrtím v prvním měsíci po LT [21], přesto byla popsána normalizace koncentrace AAT v séru již časně po transplantaci. Úspěšnost LT v 70. a 80. letech odráží tehdejší výsledky LT pro jaterní cirhózu: data z Pittsburghu z let 1980–1986 popisují 10 dospělých průměrného věku 34 let transplantovaných pro komplikace chronického selhání jater, z těchto pacientů pouze jeden trpěl CHOPN. Soubor dále zahrnoval 29 dětí, jejich průměrný věk při transplantaci byl 5 let. Pětileté přežití bylo 60 % u dospělých a 83 % u dětí a diagnóza AATD představovala tehdy 2,94 % LT u dospělých a 13 % u dětí v pittsburském transplantačním centru [22]. Data z Velké Británie jsou podobná, s jednoročním přežitím 73 % u dospělých a 87,5 % u dětí [23]. K dalšímu zlepšení výsledků LT došlo díky pokrokům v imunosupresi, chirurgických technikách a využití „splitů“ štěpů jater u dětí. Heterogenní a mnohdy nepředvídatelný klinický průběh onemocnění, zejména u dětí, kladl důraz na správné načasování zařazení pacientů do čekací listiny k LT. Indikací k LT byly zvýšené hodnoty bilirubinu a zhoršení koagulačních testů, později též hypalbuminemie, ascites a variceální krvácení [24].
Retrospektivní data z King’s College v Londýně popisují skupinu 97 dětí s AATD sledovaných po dobu 10 let: 24,7 % dětí podstoupilo LT, 73,3 % žilo bez nutnosti LT. Doba od prvních příznaků do projevů jaterního selhání byla 2,5 roku u pacientů s neonatální žloutenkou, u nemocných s projevy chronického onemocnění jater pak 4,5 roku. Přežití nemocných po LT bylo 96 % v prvním roce a 92 % 5 let po transplantaci [25]. Faktory, které predikovaly nutnost transplantace, byly novorozenecká žloutenka trvající více než 6 týdnů, vyšší hodnota AST při stanovení diagnózy a vyšší hodnoty AST a GGT v 6 měsících, v 1 roce a v 5 letech sledování. Transplantace byla rovněž častější u dětí s těžkou fibrózou a cirhózou. Celkové přežití všech dětí v souboru bylo 98 %, dvě z dětí zemřely po LT [25].
Transplantace jater u dětí
Jaterní onemocnění má u většiny dětí pomalý průběh i přes přítomnost cirhózy a portální hypertenze. U dětských pacientů se závažnou cholestázou v kojeneckém věku může dojít ke zlepšení jaterní funkce bez nutnosti LT. Transplantace by měla být indikována u pacientů s portální hypertenzí, s krvácením z jícnových varixů a s progredující jaterní dysfunkcí [26]. Portální hypertenze je u dětí s AATD častou komplikací (29 %) [27].
Data amerického transplantačního registru UNOS analyzovala výsledky LT u dětí transplantovaných mezi roky 1995 a 2004, pro AATD bylo provedeno 161 LT, 3,51 % ze všech dětských LT, průměrný věk transplantovaných dětí byl 3 roky (rozmezí 0,5–17 let). Přežití pacientů bylo excelentní: 84, 81 a 78 % jeden rok, tři roky a pět let po LT [28]. Dlouhodobé přežití 35 dětí bylo popsáno též autory z Jižní Karolíny. Na přežití měl jednoznačný vliv použitý imunosupresivní režim, u pacientů transplantovaných v době používání cyklosporinu A bylo pětileté přežití 76,5 %, zatímco nemocní léčení takrolimem dosáhli 5letého přežití ve 100 % případů [29].
Transplantace jater u dospělých
Jak bylo popsáno výše, ve středu zájmu jsou děti s AATD, u dospělých s AATD je obvykle dominantním příznakem plicní postižení. Nicméně mnoho dospělých pacientů může zůstat nediagnostikovaných či jim může být mylně přiřknuto jiné jaterní onemocnění s obdobnou klinickou manifestací, nejčastěji alkoholická choroba jater nebo nealkoholická steatohepatitida. Studie provedená na 1 953 jedincích s AATD popsala pouze 47 (2,4 %) nemocných, u nichž byla provedena LT [30], přestože jaterní cirhóza představuje významnou příčinu morbidity a mortality pacientů s AATD. V americkém NHLBI registru představovala cirhóza příčinu 10 % všech úmrtí a byla základní příčinou úmrtí u 28 % Pi*ZZ homozygotů, kteří nekouřili [17,31].
Americký transplantační registr UNOS popisuje 406 LT pro AATD mezi roky 1995 a 2004, což představovalo 1,06 % všech LT provedených u dospělých pacientů. Rizikovým faktorem nutnosti transplantace byl vyšší věk a mužské pohlaví. Muži představovali 72 % transplantovaných pacientů, průměrný věk při transplantaci byl 52 let (rozmezí 18–70 let). Přežití pacientů bylo vynikající, 89, 85 a 83 % jeden rok, tři roky a pět let po transplantaci. Pouze tři pacienti podstoupili před LT rovněž transplantaci plic. Limitací této retrospektivní studie je absence genetického vyšetření (mohlo jít nejen o Pi*ZZ, ale i o Pi*MZ nemocné), rovněž nebyly k dispozici údaje o komorbiditách, jako jsou nadměrná konzumace alkoholu, obezita, diabetes mellitus či infekce virem hepatitidy C [28].
Recentní analýza z databáze OPTN (Organ Procurement and Transplantation Network) a dvou dalších amerických transplantačních registrů identifikovala jako nejčastější období nutnosti transplantace věk mezi 50 a 64 roky, muži byli ve větším riziku transplantace než ženy. Od roku 2002 do roku 2012 představovali dospělí 87 % všech pacientů transplantovaných pro AATD [32].
Nejlépe charakterizovanou kohortou pacientů je retrospektivní analýza ze tří transplantačních center v USA, pacienti podstoupili LT mezi roky 1987 a 2002. Do analýzy bylo zařazeno celkem 73 pacientů, 50 Pi*ZZ homozygotů a Pi*SZ složených heterozygotů [33]. Skupina Pi*MZ heterozygotů byla použita jako skupina kontrolní. Muži středního věku potřebovali LT nejčastěji. LT pro AATD představovala 1,4 % všech LT, naprostá většina transplantovaných byli dospělí (97 %), jejich 5leté přežití bylo 82 %. Pouze jeden pacient zemřel na plicní komplikace více než 9 let po LT. Z analýzy rovněž vyplývá, že u mnoha pacientů bylo diagnostikováno současně jiné jaterní onemocnění (u 8 % Pi*ZZ homozygotů a u 43,4 % Pi*SZ složených heterozygotů). Toto zjištění podtrhuje význam testování AATD u všech pacientů s jaterní cirhózou [33].
Dostupná literatura zabývající se transplantací pro AATD má mnoho limitací. Onemocnění má široké spektrum klinické symptomatologie a ve většině případů se jedná o retrospektivní studie na malých skupinách pacientů, které jsou často nedostatečně geneticky definované (Pi*ZZ i Pi*SZ nemocní), s různě vysokým rizikem rozvoje onemocnění jater. Mnohdy se též jedná o nemocné zařazené na základě histologického nálezu PAS-D pozitivních granul v jaterní biopsii či jaterním explantátu, což samo o sobě není dostatečné ke stanovení diagnózy AATD, do studie mohou být tak mylně zařazeni i Pi*MZ heterozygoti.
Indikace k LT má být zvážena u všech pacientů s AATD a konečným stadiem selhání jater, u nichž předpokládáme, že jim LT prodlouží život ve srovnání s přirozeným průběhem onemocnění nebo jim významně zlepší kvalitu života [34]. O LT máme uvažovat, pokud předpokládáme, že přežití pacienta by bylo kratší než jeden rok. Pacient má postoupit podrobné vyšetření k vyloučení komorbidit, které by LT znemožnily. Nejčastějším důvodem LT u pacientů s AATD je chronické jaterní selhání při cirhóze. Tito nemocní mají být odesláni do transplantačního centra v okamžiku vzniku závažných komplikací, jako jsou variceální krvácení, ascites, encefalopatie, hepatorenální syndrom. U pacientů s HCC je LT optimální léčebnou metodou v případě časného, neresekabilního tumoru. Pacienti splňující tzv. Milánská kritéria (1 ložisko HCC do 5 cm nebo 3 ložiska HCC do 3 cm) dosahují excelentního přežití (5 let ve více než 70 % případů) [35].
Načasování LT je zcela zásadní, LT má být provedena dříve, než pacient vyvine život ohrožující komplikace jaterního onemocnění. K posouzení pokročilosti jaterního onemocnění je možno použít Child-Pughovo skóre nebo skóre MELD (Model of End-Stage Liver Disease) [36]. LT má být zvažována u pacientů Child-Pughovy klasifikace B a C a MELD skóre 15 a vyšším [34].
AATD a riziko HCC
Riziko HCC a jeho prevalence u nemocných s AATD je vysoce variabilní a závisí na studované populaci pacientů [4,37]. Ve švédské studii publikované v roce 1996, založené na analýze autoptických nálezů, bylo relativní riziko HCC 5násobné (95% konfidenční interval 1,6–15,8). Data z registru UNOS (United Network of Organ Sharing) zahrnující 567 transplantovaných pacientů s AATD nepopisují ani jeden případ HCC [28], naopak studie E. Careyové popisuje prevalenci vysokou, mezi 10 a 19 % [33]. V každém případě je incidence HCC u pacientů s AATD nižší než u jiných diagnóz vedoucích k jaterní cirhóze, jako jsou např. HCV či HBV infekce, roční kumulativní riziko pro AATD je 0,88 % (pro HCV pak 2,7 %) [38].
Vyšetření plicních funkcí před LT u pacientů s AATD
Pacienti s AATD mají riziko plicního emfyzému, což může zásadně ovlivnit možnost podstoupit LT, pokud by plicní postižení bylo závažné. CHOPN při AATD se obvykle projeví již po 30. roce věku, nepředstavuje tedy kontraindikaci LT u dětí. U dospělých nejsou k dispozici jednoznačná doporučení, kdy již pokročilost plicního onemocnění LT brání [39]. Plicní funkce může být u jednotlivých pacientů velmi rozdílná, od normálních hodnot po těžkou obstrukční poruchu, zejména u kuřáků. Progrese plicního onemocnění je na jaterním onemocnění zcela nezávislá. V každém případě by kandidáti LT měli podstoupit podrobné funkční pneumologické vyšetření, to však může být limitováno přítomností ascitu, hydrothoraxu, malnutricí či spoluprací nemocného v průběhu vyšetření při jaterní encefalopatii.
Kombinovaná transplantace jater a plic může být metodou volby u nemocných se závažnou dysfunkcí obou orgánů, jedná se však o indikaci výjimečnou [28].
Možnost LT od žijícího dárce
Transplantace jater štěpem od žijícího dárce má být zvažována zejména u dětí i přesto, že oba rodiče budou nejpravděpodobněji Pi*MZ heterozygoty. Tito dárci mají nižší sérové koncentrace AAT ve srovnání s kontrolami, biopsie štěpu provedené po reperfuzi však prokazují normální nález v jaterní tkáni, bez cholestázy, steatózy, zánětu a nálezu PAS-D pozitivních granul. Štěpy tak mohou být bezpečně použity k transplantaci [40].
Závěr
AATD je genetické onemocnění, které při závažném klinickém průběhu může vyžadovat LT jak v dětském, tak i dospělém věku. LT představuje jedinou kurativní metodu, po LT má příjemce jaterního štěpu fenotyp dárce orgánu a normální sérové koncentrace AAT. Přežití nemocných po LT je excelentní.
Zdroje
1. Strnad P, McElvaney NG, Lomas DA. Alpha1- -Antitrypsin Deficiency. N Engl J Med 2020; 382 (15): 1443–1455. doi: 10.1056/NEJMra1910234.
2. Feldmann G, Martin JP, Sesboue R et al. The ultrastructure of hepatocytes in alpha-1-antitrypsin deficiency with the genotype Pi. Gut 1975; 16 (10): 796–799. doi: 10.1136/gut.16.10.796.
3. Remih K, Amzou S, Strnad P. Alpha1-antitrypsin deficiency: New therapies on the horizon. Curr Opin Pharmacol 2021; 59: 149–156. doi: 10.1016/j.coph.2021.06.001.
4. Rabekova Z, Frankova S, Jirsa M et al. Alpha-1 Antitrypsin and Hepatocellular Carcinoma in Liver Cirrhosis: SERPINA1 MZ or MS Genotype Carriage Decreases the Risk. Int J Mol Sci 2021; 22 (19). doi: 10.3390/ijms221910560.
5. Blanco I, Bueno P, Diego I et al. Alpha-1 antitrypsin Pi*Z gene frequency and Pi*ZZ genotype numbers worldwide: an update. Int J Chron Obstruct Pulmon Dis 2017; 12: 561–569. doi: 10.2147/COPD.S125389.
6. Janciauskiene S, Eriksson S, Callea F et al. Differential detection of PAS-positive inclusions formed by the Z, Siiyama, and Mmalton variants of alpha1-antitrypsin. Hepatology 2004; 40 (5): 1203–1210. doi: 10.1002/hep.20451.
7. Lomas DA, Evans DL, Finch JT et al. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992; 357 (6379): 605–607. doi: 10.1038/357605a0.
8. Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med 2009; 360 (26): 2749–2757. doi: 10.1056/NEJM cp0900449.
9. Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med 1976; 294 (24): 1316–1321. doi: 10.1056/NEJM197606102942404.
10. Bernspang E, Carlson J, Piitulainen E. The liver in 30-year-old individuals with alpha (1) -antitrypsin deficiency. Scand J Gastroenterol 2009; 44 (11): 1349–1355. doi: 10.3109/003655 20903296669.
11. DeMeo DL, Silverman EK. Alpha1-antitrypsin deficiency. 2: genetic aspects of alpha (1) -antitrypsin deficiency: phenotypes and genetic modifiers of emphysema risk. Thorax 2004; 59 (3): 259–264. doi: 10.1136/thx.2003.006502.
12. Hope PL, Hall MA, Millward-Sadler GH et al. Alpha-1-antitrypsin deficiency presenting as a bleeding diathesis in the newborn. Arch Dis Child 1982; 57 (1): 68–70.
13. Ghishan FK, Gray GF, Greene HL. alpha 1-antitrypsin deficiency presenting with ascites and cirrhosis in the neonatal period. Gastroenterology 1983; 85 (2): 435–438.
14. Odievre M, Martin JP, Hadchouel M et al. Alpha1-antitrypsin deficiency and liver disease in children: phenotypes, manifestations, and prognosis. Pediatrics 1976; 57 (2): 226–231.
15. Eriksson S, Carlson J, Velez R. Risk of cirrhosis and primary liver cancer in alpha 1-antitrypsin deficiency. N Engl J Med 1986; 314 (12): 736–239. doi: 10.1056/NEJM198603203141202.
16. Fromme M, Schneider CV, Trautwein C et al. Alpha-1 antitrypsin deficiency: A re-surfacing adult liver disorder. J Hepatol 2022; 76 (4): 946–958. doi: 10.1016/j.jhep.2021.11.022.
17. Tanash HA, Nilsson PM, Nilsson JA et al. Clinical course and prognosis of never-smokers with severe alpha-1-antitrypsin deficiency (PiZZ). Thorax 2008; 63 (12): 1091–1095. doi: 10.1136/thx.2008.095497.
18. Strnad P, Buch S, Hamesch K et al. Heterozygous carriage of the alpha1-antitrypsin Pi*Z variant increases the risk to develop liver cirrhosis. Gut 2019; 68 (6): 1099–1107. doi: 10.1136/gutjnl-2018-316228.
19. McElvaney NG, Burdon J, Holmes M et al. Long-term efficacy and safety of alpha1 proteinase inhibitor treatment for emphysema caused by severe alpha1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med 2017; 5 (1): 51–60. doi: 10.1016/S2213-2600 (16) 30430-1.
20. Strnad P, Mandorfer M, Choudhury G et al. Fazirsiran for Liver Disease Associated with Alpha (1) -Antitrypsin Deficiency. N Engl J Med 2022; 387 (6): 514–524. doi: 10.1056/NEJMoa2205416.
21. Putnam CW, Porter KA, Peters RL et al. Liver replacement for alpha1-antitrypsin deficiency. Surgery 1977; 81 (3): 258–261.
22. Esquivel CO, Vicente E, Van Thiel D et al. Orthotopic liver transplantation for alpha-1-antitrypsin deficiency: an experience in 29 children and ten adults. Transplant Proc 1987; 19 (5): 3798–1802.
23. Vennarecci G, Gunson BK, Ismail T et al. Transplantation for end stage liver disease related to alpha 1 antitrypsin. Transplantation 1996; 61 (10): 1488–1495. doi: 10.1097/000 07890-199605270-00014.
24. Prachalias AA, Kalife M, Francavilla R et al. Liver transplantation for alpha-1-antitrypsin deficiency in children. Transpl Int 2000; 13 (3): 207–210. doi: 10.1007/s001470050688.
25. Francavilla R, Castellaneta SP, Hadzic N et al. Prognosis of alpha-1-antitrypsin deficiency-related liver disease in the era of paediatric liver transplantation. J Hepatol 2000; 32 (6): 986–992. doi: 10.1016/s0168-8278 (00) 80103-8.
26. Bakula A, Socha P, Pawlowska J et al. Good and bad prognosis of alpha-1-antitrypsin deficiency in children: when to list for liver transplantation. Transplant Proc 2007; 39 (10): 3186–3188. doi: 10.1016/j.transproceed.2007.09.046.
27. Teckman JH, Rosenthal P, Abel R et al. Baseline Analysis of a Young alpha-1-Antitrypsin Deficiency Liver Disease Cohort Reveals Frequent Portal Hypertension. J Pediatr Gastroenterol Nutr 2015; 61 (1): 94–101. doi: 10.1097/MPG.0000000000000753.
28. Kemmer N, Kaiser T, Zacharias V et al. Alpha-1-antitrypsin deficiency: outcomes after liver transplantation. Transplant Proc 2008; 40 (5): 1492–1494. doi: 10.1016/j.transproceed. 2008. 02.075.
29. Hughes MG Jr., Khan KM, Gruessner AC et al. Long-term outcome in 42 pediatric liver transplant patients with alpha 1-antitrypsin deficiency: a single-center experience. Clin Transplant 2011; 25 (5): 731–736. doi: 10.1111/ j.1399-0012.2010.01371.x.
30. Strange C, Stoller JK, Sandhaus RA et al. Results of a survey of patients with alpha-1 antitrypsin deficiency. Respiration 2006; 73 (2): 185–190. doi: 10.1159/000088061.
31. Stoller JK, Tomashefski J Jr., Crystal RG et al. Mortality in individuals with severe deficiency of alpha1-antitrypsin: findings from the National Heart, Lung, and Blood Institute Registry. Chest 2005; 127 (4): 1196–1204. doi: 10.1378/chest.127.4.1196.
32. Chu AS, Chopra KB, Perlmutter DH. Is severe progressive liver disease caused by alpha-1-antitrypsin deficiency more common in children or adults? Liver Transpl 2016; 22 (7): 886–894. doi: 10.1002/lt.24434.
33. Carey EJ, Iyer VN, Nelson DR et al. Outcomes for recipients of liver transplantation for alpha-1-antitrypsin deficiency-related cirrhosis. Liver Transpl 2013; 19 (12): 1370–1376. doi: 10.1002/lt.23744.
34. European Association for the Study of the Liver. Electronic address eee. EASL Clinical Practice Guidelines: Liver transplantation. J Hepatol 2016; 64 (2): 433–485. doi: 10.1016/ j.jhep.2015.10.006.
35. Mazzaferro V, Regalia E, Doci R et al. Liver transplantation for the treatment of small hepatocellular carcinomas in patients with cirrhosis. N Engl J Med 1996; 334 (11): 693–699. doi: 10.1056/NEJM199603143341104.
36. Wiesner R, Edwards E, Freeman R et al. Model for end-stage liver disease (MELD) and allocation of donor livers. Gastroenterology 2003; 124 (1): 91–96. doi: 10.1053/gast.2003.50016.
37. Elzouki AN, Eriksson S. Risk of hepatobiliary disease in adults with severe alpha 1-antitrypsin deficiency (PiZZ): is chronic viral hepatitis B or C an additional risk factor for cirrhosis and hepatocellular carcinoma? Eur J Gastroenterol Hepatol 1996; 8 (10): 989–994. doi: 10.1097/00042737-199610000-00010.
38. Antoury C, Lopez R, Zein N et al. Alpha-1 antitrypsin deficiency and the risk of hepatocellular carcinoma in end-stage liver disease. World J Hepatol 2015; 7 (10): 1427–1432. doi: 10.4254/wjh.v7.i10.1427.
39. Krowka MJ, Wiesner RH, Heimbach JK. Pulmonary contraindications, indications and MELD exceptions for liver transplantation: a contemporary view and look forward. J Hepatol 2013; 59 (2): 367–374. doi: 10.1016/j.jhep.2013.03.026.
40. Doshi SD, Wood L, Abt PL et al. Outcomes of Living-donor Liver Transplantation Using Grafts Heterozygous for alpha-1 Antitrypsin Gene Mutations. Transplantation 2019; 103 (6): 1175–1180. doi: 10.1097/TP.0000000000002 493.
Poděkování
Děkujeme doc. MUDr. Evě Sticové, Ph.D., a doc. MUDr. Ondřeji Fabiánovi, Ph.D., za poskytnutí obrazové dokumentace.
ORCID autorů
S. Fraňková 0000-0002-1462-5920,
M. Holinka 0009-0007-2353-5189,
J. Šperl 0000-0001-8619-2610.
Štítky
Dětská gastroenterologie Gastroenterologie a hepatologie Chirurgie všeobecnáČlánek vyšel v časopise
Gastroenterologie a hepatologie

2024 Číslo 2
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Horní limit denní dávky vitaminu D: Jaké množství je ještě bezpečné?
- Léčba akutní pooperační bolesti z pohledu ortopeda
- Ferinject: správně indikovat, správně podat, správně vykázat
Nejčtenější v tomto čísle
- Vývoj indikací k transplantaci jater
- Transplantace jater pro chronickou hepatitidu C – význam protivirové léčby v roce 2024
- Transplantace jater pro deficit alfa-1-antitrypsinu
- Diagnostika alkoholovej choroby pečene u gastroenterológa