
Hereditární renální nádorové syndromy
Authors:
Jiří Kolář 1; Tomáš Pitra 1; Kristýna Pivovarčíková 2; Radka Jaklová 3; Tomáš Zavoral 3; Ivan Trávníček 1; Hana Sedláčková 1; Kristýna Procházková 4; Tomáš Vaneček 2; Ondřej Hes 2; Milan Hora 1
Authors‘ workplace:
Urologická klinika LF UK a FN Plzeň
1; Šiklův ústav patologie LF UK a FN Plzeň
2; Ústav lékařské genetiky LF UK a FN Plzeň
3; Chirurgická klinika LF UK a FN Plzeň
4
Published in:
Ces Urol 2020; 24(1): 26-41
Category:
Review article
Overview
V posledních letech dochází ke zvyšování počtu definovaných hereditárních nádorových syndromů, z nichž některé jsou spojeny s predispozicí k rozvoji renálního karcinomu. Na hereditární renální nádorový syndrom je třeba pomýšlet u mladých jedinců s renálním karcinomem, u pacientů s pozitivní rodinnou anamnézou nádorů ledvin, dále u renálních lézí bilaterálních a multifokálních a při výskytu extrarenálních symptomů charakteristických pro některý ze syndromů. Mezi nejčastější hereditární nádorové syndromy asociované s renálním karcinomem patří syndrom von Hippel‑Lindau, Birt‑Hogg‑Dubé syndrom, hereditární papilární renální karcinom, hereditární leiomyomatóza a renální karcinom, sukcinátdehydrogenáza deficientní renální karcinom, komplex tuberózní sklerózy a Cowdenův syndrom. V souvislosti s hereditárním renálním karcinomem jsou popsány mutace v tumor supresorových genech (VHL, BAP1, PTEN, TSC1, TSC2) i v protoonkogenech (MET). V rámci jednotlivých syndromů se setkáváme s rozdíly ve stupni penetrance onemocnění, v histologii jednotlivých typů renálního karcinomu, v jeho agresivitě a metastatickém potenciálu, a tedy i v následné léčbě a dispenzarizaci.
Klíčová slova:
genetika – hereditární syndrom – renální karcinom – surveillance – zárodečná mutace
ÚVOD
Většina renálních karcinomů (RCC – Renal Cell Carcinoma) vzniká sporadicky, hereditární nádory ledvin tvoří dle literatury 3–8 % všech renálních tumorů (1, 2). V rámci výzkumného programu „the Cancer Genome Atlas (TCGA)“, v němž byly analyzovány mimo jiné i zárodečné mutace u RCC, byla u 6 % případů světlobuněčného renálního karcinomu z celkového počtu 387 prokázána zárodečná patogenní mutace; u papilárního renálního karcinomu byla zárodečná patogenní varianta prokázána v 9 % a u chromofóbního renálního karcinomu pak v 6 % (3). V posledních letech dochází ke zvyšování počtu popsaných hereditárních syndromů, z nichž některé jsou spojeny s predispozicí k RCC. V roce 2016 byly do WHO klasifikace nádorů ledvin zařazeny podjednotky asociované s genetickými syndromy – sukcinátdehydrogenáza (SDH)-deficientní RCC a renální karcinom asociovaný se syndromem hereditární leiomyomatózy. Incidence renálního karcinomu, podobně jako u ostatních malignit, stoupá s věkem. Medián věku stanovení diagnózy RCC je 64 let, naproti tomu u hereditárních nádorových syndromů je téměř o dvě a půl dekády nižší (4). Mezi nejčastější dědičné nádorové syndromy asociované s renálním karcinomem patří syndrom von Hippel‑Lindau (VHL), hereditární papilární renální karcinom (HPRCC), hereditární leiomyomatóza a renální karcinom (HLRCC), syndrom Birt‑Hogg‑Dubé (BHD), SDH asociovaný renální karcinom, komplex tuberózní sklerózy (TSC) a Cowdenův syndrom. Diagnostika a následná léčba renálního karcinomu v rámci dědičného syndromu se liší od onemocnění sporadického. Důležité je v první řadě na možnost hereditárního syndromu vůbec pomyslet, což má pak vliv nejen na prognózu pacienta, ale i na včasný záchyt onemocnění u členů rodiny, kteří mohou být onemocněním také postiženi. Familiární renální karcinom je zpravidla dědičný autosomálně dominantně (někdy s neúplnou penetrancí), nicméně jedinci s mutací predisponujícího genu nemusí nutně mít rodinnou anamnézu RCC (mutace může u probanda vzniknout de novo nebo se u rodičů neprojevila pro neúplnou penetranci). Na hereditární renální karcinom je třeba myslet, zejména pokud se jedná o RCC u pacientů mladých (≤ 46 let), pokud jsou renální léze bilaterální či multifokální nebo pokud se u pacienta vyskytují extrarenální symptomy charakteristické pro jednotlivé syndromy (například cerebelární hemangiblastomy u VHL, kožní angiofibromy u tuberózní sklerózy, plicní cysty u BHD či kožní a děložní leiomyomy u HLRCC). U této skupiny pacientů bychom měli doplnit genetické vyšetření, k jehož indikaci mohou přispět i specifické histopatologické znaky či výsledky imunohistochemického vyšetření (5). Stupeň penetrance renálního karcinomu se liší mezi jednotlivými syndromy, stejně jako agresivita tumoru, jeho léčba a následná dispenzarizace. Genetické vyšetření, k němuž se nejčastěji využívají lymfocyty z periferní krve, spočívá v průkazu zárodečné mutace. Po izolaci DNA probíhá vyšetření nádorového panelu s využitím metod masivně paralelního sekvenování (sekvenování nové generace, Next Generation Sequencing – NGS) s následným doplněním vyšetření MLPA (Multiplex Ligation‑dependent Probe Amplification), které slouží k vyšetření velkých delecí a duplikací, které nemusí být při NGS zachyceny. U nemocných s prokázaným hereditárním nádorovým syndromem je pak možné využít při plánování potomka metod preimplantační diagnostiky (PGD – Preimplanatation Genetic Diagnosis), kdy je při umělém oplodnění (IVF) cíleně vyšetřena nádorová mutace před implantací blastomery do dělohy a lze tak zabránit přenosu dané choroby do další generace.
Fig. 1. CT scan, transverse plane. Thirty-seven year old
male with a VHL syndrome. 18mm tumor in the ventral
portion of the solitary kidney. The laparoscopy-assisted
radiofrequency ablation was performed in 2006. Before
that, multiple kidney surgeries were done because of
the ccRCC (left transperitoneal nephrectomy, 2x partial
nephrectomy of the right kidney, extirpation of the metastasis of the ccRCC from left retroperitoneum). Status
post left inguinal orchiectomy (papillary cystadenoma
of the epididymis). Status post extirpation of the hemangioblastoma of the cerebellum

Tab. 1. Overview of major hereditary renal cell cancer syndromes

Fig. 2. CT scan, coronal plane. Forty-five year old male
with a VHL syndrome. Multifocal bilateral ccRCC (verified
by biopsy) with metastases (adrenal gland, pancreas,
suspected lymph node involvement). Status post extirpation of the hemangioblastoma of the cerebellum. Palliative
targeted therapy with Sunitinib is being administered with
significant partial regression of the tumor and metastases.
Patients from fig. 1 and fig. 2 are siblings

SYNDROM VON HIPPEL LINDAU
Syndrom von Hippel‑Lindau (VHL) byl prvním definovaným nádorovým syndromem asociovaným s renálním karcinomem. Jedná se o autosomálně dominantně (AD) dědičné onemocnění s téměř kompletní penetrancí do věku 65 let a s incidencí 1 : 36000. Příčinou je zárodečná mutace tumor supresorového genu VHL (3p25.3) (6). Produkt VHL genu – protein VHL (pVHL) je ligáza, která při normoxii ubikvitinuje hypoxický inducibilní faktor 1α (HIF1α) a tak způsobí jeho degradaci v proteazomu. Při mutaci VHL HIF1α uniká degradaci a po heterodimerizaci s β podjednotkou aktivuje řadu růstových, angiogenních a mitogenních faktorů (VEGF, PDGFβ, TGFα) (7). Spektrum klinických projevů syndromu VHL je široké, kromě RCC nejčastěji vznikají hemangioblastomy mozečku, mozkového kmene, míchy a sítnice (až u 70 % nemocných), méně frekventované jsou feochromocytomy/paragangliomy (20 % nemocných), pankreatické neuroendokrinní tumory (přibližně 10 % nemocných) či nádory endolymfatického vaku (5–10 % nemocných). Dále se mohou objevit cysty v různých lokalizacích (ledviny, pankreas, játra, slezina) a cystadenom nadvarlete či širokého vazu děložního. Pokud bychom postižení jednotlivých orgánů srovnali dle věku pacienta do časové osy, hemangioblastom sítnice je diagnostikován v průměrném věku 25 let (1–67), postižení CNS ve 30 letech (11–78) a RCC v 37 letech (16–67) (8, 9). Závažnost renálního postižení je variabilní, dokonce i mezi členy postižené rodiny. U některých nemocných s VHL renální karcinom nikdy nevznikne, jiní mají jen malé množství renálních cyst a další mohou mít bilaterální multifokální renální karcinom se stovkami renálních lézí. Syndrom VHL se rozděluje do pěti subtypů dle typu mutace a tumorózního spektra. Riziko RCC je nejvyšší u typu IA a IIB (1, 10). Průměrný věk diagnózy symptomatického RCC je 37 let (16–67 let) (11, 12). Riziko vzniku renálního karcinomu u nemocných s VHL je přes 70 % (13). Histologicky se v drtivé většině případů jedná o světlobuněčný renální karcinom (ccRCC), vzácně se může vyskytovat světlobuněčný papilární karcinom (14). Vzhledem k multiorgánovému postižení je u nemocných s VHL nutný interdisciplinární přístup. Stran viscerálních manifestací VHL se u asymptomatických jedinců doporučuje USG břicha jednou ročně se začátkem v 8 letech věku, od 15 let se doporučuje jednou ročně provést CT nebo MRI břicha (15). Dále je nutná dispenzární péče oftalmologa (roční kontroly), neurologa (MRI mozku a míchy v intervalech 1–2 roky se začátkem v 11 letech), každoroční audiologické vyšetření a pravidelné roční kontroly metanefrinů v plazmě nebo v moči. Vzhledem k multifokalitě a tendenci k recidivám se doporučuje u renálních lézí do 3 cm iniciálně pečlivé aktivní sledování a pokud velikost léze překročí 3 cm poté zasáhnout, s preferencí ledvinu šetřících výkonů (resekce, radiofrekvenční ablace). Walther et al. nezaznamenali během pětiletého sledování žádnou metastázu ani potřebu transplantace ledviny či dialýzy u 52 pacientů s tumorem < 3 cm. Naopak, vzdálené metastázy se objevily u 11 ze 44 nemocných (25 %) s lézemi > 3 cm, včetně 3 z 27 pacientů (11 %) s velikostí tumoru 3–6 cm (16). Podobné výsledky měl Duffey et al. pět let poté (17). Toto „pravidlo 3 cm“ je využíváno nejen u syndromu VHL, ale i u pacientů s BHD a HPRCC.
SYNDROM TUBERÓZNÍ SKLERÓZY
Komplex tuberózní sklerózy (TSC) je AD dědičné genetické onemocnění s incidencí přibližně 1 : 10000, při kterém může být postižen prakticky jakýkoliv orgánový systém. Příčinou onemocnění je inaktivační mutace v tumor supresorových genech TSC1 (9q34, kódující protein hamartin) nebo TSC2 (16p13.3, kódující protein tuberin). Pro onemocnění je typická inter - i intrafamiliární variabilita symptomů i závažnost postižení. Penetrance TSC je asi 95 %, přičemž 65–85 % případů je projevem mutace de novo (18, 19). Diagnóza TSC může být stanovena při splnění klinických diagnostických kritérií (tab. 2) nebo na základě genetického vyšetření, které indikuje klinický genetik při podezření na onemocnění, a u příbuzných osob TSC pacientů, kteří mají riziko nosičství mutace TSC genu (20, 21). U nemocných s TSC bývá zpravidla postižen nervový systém (hamartomy mozku a sítnice, subependymální obrovskobuněčné astrocytomy). Z neurologického postižení plynou charakteristické symptomy TSC souhrnně označované jako TAND (TSC‑Associated Neuropsychiatric Disorder) tj. epilepsie, mentální retardace, autismus apod. Dále bývají postiženy plíce (plicní lymfangiomyomatóza), kůže (faciální angiofibromy – adenoma sebaceum, suba periunguální fibromy, hypomelanotické makuly, šagrénové skvrny) a srdce (rhabdomyom). U 50–80 % nemocných s tuberózní sklerózou nacházíme renální angiomyolipomy (často multifokální a bilaterální) a přibližně u poloviny cystické postižení ledvin (18, 22). U pacientů s TSC nacházíme jak klasické, tak vzácně i potencionálně maligní epiteloidní angiomyolipomy, jejichž incidence je u pacientů s TSC vyšší než u běžné populace. Navzdory benignímu chování angiomyolipomů mohou problémy činit lokální symptomy u velkých lézí, riziko vzniku renálního selhání a ruptury s následným až život ohrožujícím krvácením. Vaskulární složka angiomyolipomu často obsahuje cévní aneuryzmata, riziko ruptury a následné hemoragie se signifikantně zvyšuje u aneuryzmat větších než 5mm a u lézí nad 4 cm (22). Renální karcinom vzniká u 2–4 % pacientů s tuberózní sklerózou, průměrný věk diagnózy RCC u TSC pacientů je 28 let (23). Histologicky se může jednat o renální karcinom s hlad ‑ kosvalovým stromatem, onkocytom, chromofobní renální karcinom nebo eosinofilní, solidní a cystický RCC (ESC RCC) (18, 24, 25). ESC RCC je nedávno popsaná jednotka, která většinou vzniká sporadicky, ale přibližně v 10 % případů se vyskytuje právě u pacientů s TSC (26, 27). Ve srovnání se sporadickým RCC, TSC asociované renální karcinomy mají specifické klinicko‑patologické znaky zahrnující mladší věk diagnózy, predominanci ženského pohlaví, vícečetné RCC (synchronní i metachronní), asociaci s angiomyolipomy a indolentní chování (18). Vyšší riziko AML a renálních cyst mají pacienti s onemocněním, které vzniklo de novo, a pacienti s TSC2 mutací (28). Při dispenzarizaci pacientů s TSC je vzhledem k multiorgánovému postižení, podobně jako u syndromu VHL, nutná interdisciplinární spolupráce. Stran renálního postižení je doporučeno provádět MRI břicha v intervalech 1–3 roky se začátkem v době diagnózy TSC. MRI je preferováno před USG pro vyšší senzitivitu záchytu angiomyolipomů s minimálním obsahem tuku a renálního karcinomu (11). AML s malým podílem tukové složky nacházíme asi u třetiny pacientů s TSC, odlišení od renálních tumorů včetně RCC je často možné až na základě provedení biopsie tohoto ložiska. Stran dalšího sledování pacientů s TSC by se měl v pravidelných ročních intervalech kontrolovat krevní tlak a vyšetřovat renální funkce. Mezi indikace k chirurgickému výkonu patří významné retroperitoneální krvácení, lokální symptomy (perzistující bolest) a suspektní přítomnost renálního karcinomu. Opakované resekce renálních angiomyolipomů jsou nahrazovány embolizací přívodných cév. Inaktivace genu TSC vede ke zvýšení exprese savčího receptoru rapamycinové dráhy. Bylo zjištěno, že deriváty rapamycinu – mTOR inhibitory (everolimus, sirolimus) – mají inhibiční účinek na produkci VEGF, růst nádoru a angiogenezi (29, 30, 31). Byla provedena mezinárodní, dvojitě zaslepená studie (EXIST II), která prokázala významné snížení objemu nádoru – v průměru po osmiměsíčním podáváním everolimu došlo u 42 % pacientů (33/79) k více než 50% zmenšení objemu angiomyolipomů (32, 33, 34). V roce 2017 vyšel update studie EXIST II, jehož cílem bylo zhodnotit efekt a eventuální rizika a nežádoucí účinky dlouhodobé terapie mTOR inhibitory. Výsledkem této studie bylo, že čtyřleté používání everolimu zabraňuje zvětšování angiomyolipomů ledvin nebo tento proces zpomaluje, zabraňuje poklesu renální funkce, snižuje nutnost invazivních výkonů a je relativně bezpečné (33). mTOR inhibitory jsou schváleny k léčbě dospělých pacientů s TSC s renálním AML, kteří nevyžadu ‑ jí bezprostřední chirurgický výkon, a dospělých a dětí s TSC od 3 let věku se SEGA (SubepEndymal Giant cell Astrocytoma), kteří vyžadují terapeutický zákrok, ale nejsou kandidáty kurativní chirurgické resekce. Cílená systémová terapie poskytuje alternativu chirurgické léčby či umožňuje kurativní chirurgický výkon po redukci objemu AML (35, 36).
Tab. 2. Clinical criteria for the diagnosis of tuberous sclerosis complex

Fig. 3. The family tree of the family with Von Hippel Lindau syndrome

Fig. 4. Papillary cystadenoma of the epididymis. Patient with a VHL syndrome. H&E. Viewed at 400× magnification

Fig. 5. Clear cell renal cell carcinoma, patient with a VHL
syndrome. H&E. Viewed at 400× magnification

Fig. 6. Thirty-eight year old female with a VHL syndrome. Kidneys after transperitoneal bilateral nephrectomy,
histologically multiple clear cell renal cell carcinomas

Fig. 7. CT scan, coronal plane. Patient with tuberous
sclerosis. Kidneys with multiple angiomyolipomas and
cysts, a stripe of haemorrhage in the left retroperitoneum

Tab. 3. Referral criteria for genetic counselling

Fig. 8. Skin lesions of the patient with tuberous sclerosis. Facial angiofibromas (A) and a shagreen patch (B) in its
typical lower back localization
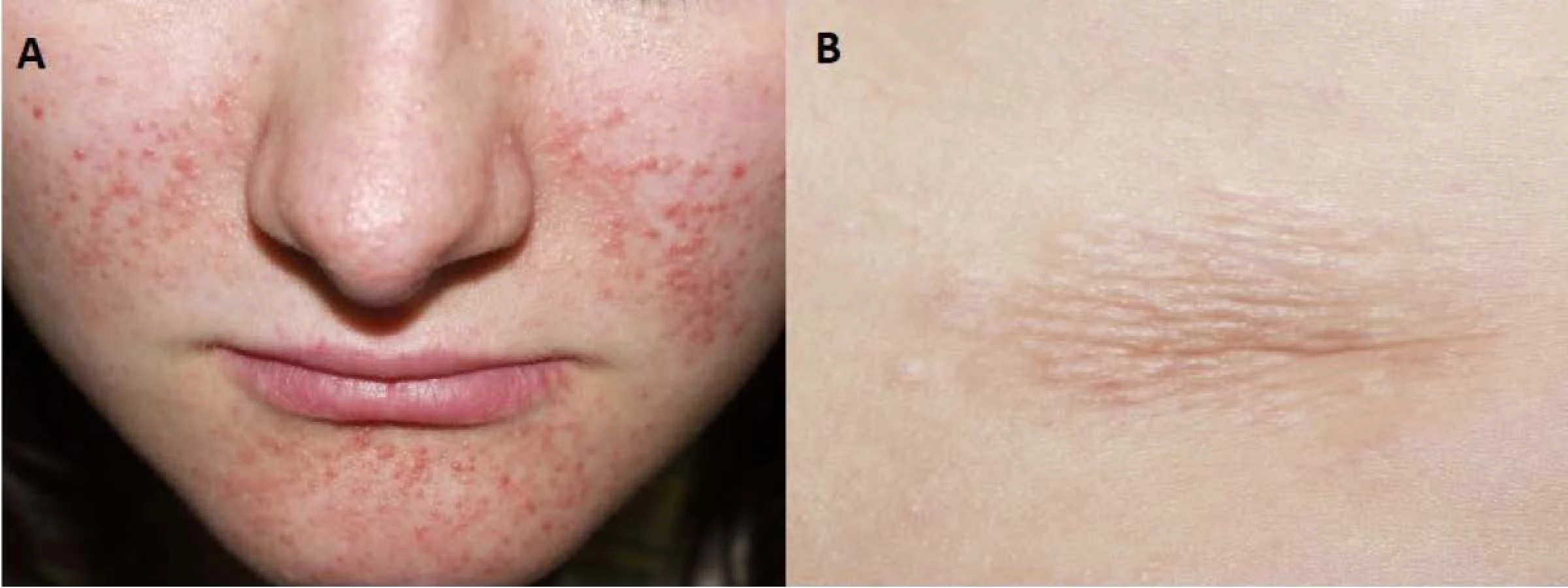
Fig. 9. MRI scan, coronal plane. Polycystic kidneys of
the patient with tuberous sclerosis

Fig. 10. Kidneys after bilateral transperitoneal nephrectomy. Histologically multiple papillary renal cell cancers
type 1 and multiple papillary adenomas, highly suspicious of HPRCC, molecular-genetic examination to confirm
MET mutation has not been yet conducted

HEREDITÁRNÍ LEIOMYOMATÓZA A RENÁLNÍ KARCINOM
Syndrom hereditární leiomyomatózy a renálního karcinomu (HLRCC, Reedův syndrom) je relativ ‑ ně vzácné (0,05–0,2 % všech renálních karcinomů) AD dědičné onemocnění s predispozicí ke tvorbě mnohočetných kožních a děložních leiomyomů a ke vzniku renálního karcinomu. Je popisováno i zvýšené riziko kožních a děložních leiomyosarkomů. Genetickým podkladem je zárodečná mutace FH genu (1q42–43), který kóduje enzym fumarát hydratázu katalyzující konverzi fumarátu na malát v Krebsově cyklu. Tato mutace vede ke zvýšení nitrobuněčné koncentrace fumarátu, který reaguje s proteiny cysteinového typu za vzniku stabilního S-(2-succino)-cysteinu (2SC). V konečném důsledku dochází k narušení procesu oxidativní fosforylace a akumulaci onkometabolitů predisponujících ke vzniku nádoru (24). HLRCC asociovaný RCC byl jako předpokládaný dědičný typ papilárního renálního karcinomu typu II zařazen již ve 2004 WHO klasifikaci renálních neoplazií, jako samostatná jednotka byl uznán v roce 2013 (International Society o Urological Pathology (ISUP) Vancouver Classification of renal tumors) a v 2016 WHO klasifikaci renálních neoplazií (37, 38). HLRCC asociovaný RCC vzniká u 10–30 % jedinců se syndromem hereditární leiomyomatózy (39). Standardní histopatologické vyšetření s hematoxylin‑eosinem není k určení diagnózy dostatečné. Pomoci může imunohistochemické vyšetření (anti-2SC, anti‑FH), pro po ‑ tvrzení diagnózy je nutná mutační analýza genu pro fumarát hydratázu (40). Renální karcinom vyskytující se u syndromu HLRCC je nádor agresivní, s vysokou letalitou, postihuje mladší jedince obou pohlaví, s lehkou převahou pohlaví mužského (40). Obvykle bývá unilaterální a solitární. Medián věku diagnózy je 39 let (24). Metastatický potenciál není přímo úměrný velikosti nádoru, metastazovat mohou i malé tumory (24). Termín FH deficientní RCC (FHRCC) se používá pro nádory se sugestivním morfologickým nálezem (multiplicita morfologického vzhledu, objemná eosinofilní makrojadérka s perinukleolárním projasněním) a typickým imunohistochemickým profilem (FH negativita, 2-SC pozitivita), avšak syndrom jako takový není vyvinut nebo znám a výsledky molekulárně genetického vyšetření nejsou jednoznačně interpretovatelné. Termínem FHRCC také můžeme označit nádory se somatickou mutací FH genu (41). Není jednotný konsenzus stran algoritmu dispenzarizace u nosičů mutace FH genu, obecně se doporučuje MRI břicha jednou ročně se začátkem v 18 letech, ultrasonografické vyšetření nemá dostatečnou senzitivitu k zachycení malých lézí. Zob ‑ razovací vyšetření břicha před 18. rokem by mělo být diskutováno s pacientem, i vzhledem k relativně nízkému riziku RCC před 20. rokem věku (kolem 1–2 %) (11). Některými autory je doporučováno sonografické vyšetření v půlročních intervalech u nosičů FH mutace se začátkem v 8–10 letech, v 18 letech pak přejít na dispenzarizaci pomocí MRI (42). Pokud je diagnostikován i jen malý karcinom ledviny v rámci HLRCC syndromu, aktivní sledování není doporučováno, ale je indikována radikální chirurgická intervence (nefrektomie či resekce se širokým okrajem). Radikální přístup u HLRCC se liší od většiny hereditárních nádorových syndromů asociovaných s RCC (VHL, HPRCC, BHD), kde jsou metodou volby nefron šetřící metody či možnost aktivního sledování u drobných ložisek. U těchto syndromů je cí ‑ lem zachování co největšího množství zdravé tkáně a z dlouhodobého hlediska renálních funkcí (43).
Fig. 11. Electropherogram of the part of 1 gene of VHL with a determined pathogenic variant c302T>G in a heterozygous state

HEREDITÁRNÍ PAPILÁRNÍ RENÁLNÍ KARCINOM
Hereditární papilární renální karcinom je AD dědičný, vzácný syndrom s vysokou penetrancí (téměř 100 % do 80 let věku), který je charakterizovaný výskytem papilárního renálního karcinomu typu 1 (PRCC-1) a papilárních renálních adenomů, typicky s multifokálním a bilaterálním postižením ledvin někdy až stovkami či tisícovkami tumorů (28). Histologicky jsou karcinomy u syndromu HPRCC neodliši ‑ telné od sporadického PRCC-1, na syndrom musíme myslet při vícečetném postižení a/nebo při pozitivní rodinné anamnéze stran RCC (44). Extrarenální manifestace nejsou popsány (24). Příčinou syndromu HPRCC je aktivační mutace protoonkogenu MET (7q31), jehož produktem je tyrosinkinázový receptor c‑MET pro hepatocytární růstový faktor. Vzhledem k absenci extrarenálních manifestací je screening omezen na ledviny. Tumory u HPRCC syndromu rostou pomalu, průměrně o 0,19 cm/rok, přesto tyto nádory mohou metastazovat (45, 46). Medián věku diagnózy HPRCC spadá do 6. dekády života, ale jsou i případy vyskytující se před 20. rokem věku (47). Stran léčby se u renálních tumorů u pacientů s HPRCC zpravidla využívá „pravidla 3 cm“. U pokročilého onemocnění vedla identifikace aktivační MET mutace u pacientů s HPRCC a se sporadickým PRCC-1 ke klinickým studiím s využitím inhibitorů MET. Byl hodnocen efekt foretinibu (inhibitor MET, VEGFR2, RON a AXL) u pacientů s PRCC. Největší benefit byl pozorován u pacientů se zárodečnou patogenní variantou MET; 5 (50 %) z 10 pacientů vykázalo parciální odpověď ve srovnání s 5 (9 %) z 57 pacientů se sporadickým tumorem (28). Stran dispenzarizace se doporučuje u rizikových pacientů provést v 18 letech CT/MRI břicha. Pokud je toto výchozí vyšetření bez patologie, další zobrazovací vyšetření, CT nebo MRI, by mělo být provedeno ve 30 letech, kdy se riziko RCC výrazně zvyšuje, a poté dále pokračovat v dvouročních intervalech (11).
SYNDROM BIRT‑HOGG‑DUBBÉ
Syndrom Birt‑Hogg‑Dubé (BHD) je AD dědičné onemocnění, které charakterizují mnohočetné kožní fibrofolikulomy (typicky v obličeji), plicní cysty, spontánní pneumotorax, riziko vzniku renálního karcinomu a kolorektálních tumorů (48). Onemocnění je vzácné (incidence 1 : 200000), má vysokou penetranci a velkou inter - a intrafamiliární variabilitu. Příčinou onemocnění je zárodečná mutace tumor supresorového genu FLCN (17p11.2), kódující protein folliculin, jehož funkce zatím nebyla plně objasněna, nicméně jeho mutace vede k aktivaci signální cesty mTOR (49). Riziko renálního karcinomu u BHD syndromu je významně nižší než u VHL – přibližně 25 %, průměrný věk diagnózy RCC je 50 let (50, 51). Nádory jsou u více než poloviny nemocných bilaterální nebo multifokální a jsou pomalu rostoucí. Histologicky se asi v 50 % případů jedná o hybridní chromofóbní/ onkocytární renální karcinom (v dřívější klasifikaci veden jako podtyp chromofóbního RCC), ale může se vyskytnout i světlobuněčný renální karcinom a papilární renální karcinom (52, 53). Diagnóza BHD syndromu se opírá o klinický obraz a molekulárně genetickou analýzu genu FLCN. Jedincům, kteří jsou nosiči mutace nebo mají klinickou diagnózu BHD syndromu bez prokázané mutace, se doporučuje od 20 let podstoupit v intervalech 1–3 roky CT nebo MRI břicha. USG vyšetření se nedoporučuje, protože hybridní chromofóbní/onkocytární nádory jsou často izoechogenní s okolním ledvinným parenchymem a mohou detekci uniknout (54).
SDHX ASOCIOVANÉ SYNDROMY (HEREDITÁRNÍ SYNDROM FEOCHROMOCYTOM‑PARAGANGLIOM)
Sukcinát dehydrogenáza (SDH) je mitochondriální enzymatický komplex, který se skládá ze čtyř různých podjednotek. V Krebsově cyklu katalyzuje oxidaci sukcinátu na fumarát a v dýchacím řetězci hraje roli v enzymovém komplexu II. Geny kódující podjednotky SDH jsou SDHA, SDHB, SDHC, SDHD a SDHAF2. Ačkoliv jednotlivé podjednotky jsou součástí stejného proteinového komplexu, mutace v jednotlivých genech vedou k rozdílnému klinickému fenotypu. Podle typu mutovaného genu rozlišujeme syndrom paragangliomu 1–5. Ztráta „wild type“ alely v tumoru společně se zárodečnou mutací znamená destabilizaci SDH komplexu a nefunkčnost enzymu (55). Mutace genů jsou spojeny se zvýšeným rizikem feochromocytomů nadledvin, paragangliomů (hlava, krk, hrudník, břicho), gastrointestinálních stromálních tumorů, nádorů hypofýzy, vznikem papilárního karcinomu štítné žlázy a renálního karcinomu (56). SDH deficientní renální karcinom tvoří přibližně 0,05–0,2 % všech RCC a průměrný věk v době diagnózy je 38 let (24, 57). Nejčastěji se vyskytuje renální karcinom asociovaný s mutací v podjednotce SDHB. Riziko vzniku RCC u nosičů mutace genu SDHB je zhruba 10–15 % (50). Podobně jako u syndromu hereditární leiomyomatózy mohou být SDHB deficientní renální karcinomy agresivní s tendencí k časnému metastazová ‑ ní, a to i u malých ložisek (2). U low grade renálních karcinomů jsou i vzhledem k častému bilaterálnímu nálezu a multifokalitě indikovány záchovné výkony na ledvině, u high grade neoplazií (sarkomatoidní změny, koagulační nekrózy, vysoký nukleární grade) s vysokým rizikem metastazování by měla být léčba radikálnější (58). Stran dispenzarizace je u nosičů mutace SDHB, SDHC a SDHD doporučeno od 15 let věku provedení CT nebo MRI břicha v ročních intervalech (11, 59).
COWDENŮV SYNDROM
Cowdenův syndrom je řazen mezi PTEN (phosphatase and tensin homolog) hamartomózní tumorové syndromy. Jedná se o syndrom vzácný (incidence 1 : 200 000), jehož příčinou je zárodečná mutace tumor supresorového genu PTEN (10q23.3). Typické jsou kožní projevy (trichi ‑ lemomy, akrální keratóza, papilomatózní papuly a slizniční léze), které se vyvíjí až u 99 % jedinců do 30 let. Je spojen se zvýšeným rizikem benigních a maligních tumorů štítné žlázy, prsu a endometria. Riziko renální karcinomu u nemocných s Cowdenovým syndromem je až třicetinásobně vyšší než u běžné populace (60). Histologicky se nejčastěji vyskytuje papilární a chromofóbní RCC (44). Stran dispenzarizace renálních lézí se doporučuje provést USG břicha jednou ročně se začátkem v 25 letech. BAP 1
HEREDITÁRNÍ NÁDOROVÝ SYNDROM A FAMILIÁRNÍ RCC
Jedná se o nedávno popsaný nádorový syndrom, jehož příčinou je zárodečná mutace tumor supresorového genu BAP1 (BRCA associated protein 1) (24). Je ‑ dinci se zárodečnou mutací BAP1 genu mají zvýšené riziko kožního a uveálního melanomu, mesoteliomu a renálního karcinomu s agresivním chováním. Histologicky nacházíme nejčastěji světlobuněčný renální karcinom (61, 62). Vzhledem k nedostatku dat zatím není definován protokol surveillance u nosičů BAP1 mutace, ale podobně jako u ostatních hereditárních nádorových syndromů bychom se měli vyvarovat opakované radiační zátěži, a tudíž by MRI mělo být preferováno před CT vyšetřením (24, 50).
RCC ASOCIOVANÝ SE SYNDROMEM „HYPERPARATHYROIDISM JAW TUMOR“ (HPT‑JT)
Příčinou tohoto velmi vzácného syndromu je zárodečná mutace tumor supresorového genu HRPT2/CDC73 kódující protein parafibromin. U tohoto syndromu je zvýšené riziko sekrečně aktivních adenomů nebo karcinomů příštítných tělí ‑ sek (způsobující primární hyperparatyroidismus), fibrooseózních lézí maxilly a mandibuly, renálních cyst a renálních neoplazií. Histologicky se nejčastěji vyskytuje smíšený epiteliální a stromální tumor (MEST), byl pospán i papilární renální karcinom a Wilmsův tumor (44, 63).
OSTATNÍ VZÁCNÉ HEREDITÁRNÍ SYNDROMY
Mezi další vzácné hereditární syndromy asociované s RCC můžeme zařadit familiární papilární karcinom štítné žlázy se zvýšeným rizikem papilárního RCC a renálních onkocytomů. Dále může být vzácnou příčinou familiárního RCC konstituční translokace chromozomu 3, kdy nacházíme multifokální, často bilaterální světlobuněčné karcinomy ledvin nerozeznatelné od sporadického světlobuněčného RCC; průměrný věk výskytu RCC je vyšší než u VHL. Z formálního hlediska můžeme mezi hereditární syndromy zařadit i medulární renální karcinom, protože je spojen s hereditární hemoglobinopatií – srpkovitou anémií. Vyskytuje se u černochů, nejčastěji ve 2. a 3. dekádě života. Nádor je vysoce agresivní, většinou pokročilý v době diagnózy, prognóza je extrémně špatná, nemocní většinou umírají v průběhu několika měsíců od stanovení diagnózy (24).MiTF (Microphthalmia‑associated Transcription Factor) predispozice k familiárnímu RCC je syndrom způsobený zárodečnou mutací genu MITF a je spojován s více než pětinásobně vyšším rizikem vzniku kožního melanomu a RCC (64). Tento článek se podrobně nezabývá familiárními pediatrickými renálními nádorovými syndromy, ačkoliv některé renální léze vyskytující se v rámci výše zmíněných syndromů se mohou manifestovat ve velmi mladém věku. Mezi familiární pediatrické renální nádorové syndromy můžeme zařadit familiární nefroblastom, WAGR syndrom (Wilmsův tumor, aniridie, urogenitální malformace, mentální retardace), Denys‑Drashův syndrom (Wilmsův tumor, mesangiální skleróza, pseudohermafroditismus) a Beckwith Wiedemannův syndrom (Wilmsův tumor, hemihypertrofie, makroglosie, omfalokéla, visceromegalie) (24).
ZÁVĚR
Hereditární nádorové syndromy asociované s RCC jsou způsobeny různými genetickými alteracemi a zahrnují široké spektrum klinických projevů, rozdílnou incidenci i typ RCC. Dědičné nádory ledvin tvoří dle literatury asi 5 % všech případů renálních karcinomů. Na našem pracovišti se ovšem incidence hereditárních případů pohybuje pouze kolem 1 %. Je to pravděpodobně dáno jejich poddiagnostikováním – klinik ne vždy na možnost dědičného případu myslí a potřebné genetické vyšetření není doplněno. Je proto nutná zvýšená erudice v této oblasti. Na možnost hereditárního renálního karcinomu by se mělo myslet u mladých jedinců s RCC (≤ 46 let), u pacientů s bilaterálními či multifokálními renálními lézemi, dále při pozitivní rodinné anamnéze stran RCC a při současném výskytu extrarenálních manifestacích charakteristických pro jednotlivé syndromy. U této skupiny pacientů bychom měli doplnit genetické vyšetření, k jehož indikaci mohou přispět i specifické histopatologické znaky či výsledky imunohistochemického vyšetření. S rozvojem vyšetřovacích metod a zejména s pokro ‑ kem v oblasti genetiky, včetně využívání metod sekvenování nové generace při genetickém vyšetření, dochází v posledních letech k nárůstu počtu definovaných hereditárních renálních syndromů. Surveillance u jedinců s rizikem dědičného RCC má za cíl včasný záchyt tumorů a s tím související snížení morbidity a mortality. Podrobnější poznání genetického pozadí hereditárních renálních karcinomů má potenciál nejen k jejich včasné diagnostice a popsání specifických léčebných a dispenzárních postupů, ale může vést i k objevení nových terapeutických cílů u karcinomu sporadického.
SEZNAM ZKRATEK
- AD – autosomálně dominantní
- BHD – syndrom Birt‑Hogg‑Dubé
- CCPAP RCC – světlobuněčný papilární renální karcinom
- CCRCC – světlobuněčný renální karcinom
- ESC RCC – eosinofilní, solidní a cystický renální karcinom
- FHRCC – fumarát hydratáza deficientní renální karcinom
- HIF – hypoxií indukovaný faktor
- HLRCC – hereditární leiomyomatóza a renální karcinom
- HPRCC – hereditární papilární renální karcinom
- CHRCC – chromofóbní renální karcinom
- LAM – lymfangiomyomatóza
- MEST – smíšený epiteliální a stromální tumor
- MITF – transkripční faktor asociovaný s mik ‑ roftalmií
- MTOR – Mammalian Target Of Rapamycin
- NET – neuroendokrinní tumor
- NGS – sekvenování nové generace
- PDGF – destičkový růstový faktor
- PRCC – papilární renální karcinom
- PTEN – Phosphatase and Tensin Homolog
- PVHL – protein VHL
- SDH – sukcinát dehydrogenáza
- TCGA – The Cancer Genome Atlas
- TGF – transformující růstový faktor
- TSC – komplex tuberózní sklerózy
- VHL – syndrom Von Hippel Lindau
- VEGF – vaskulární endoteliální růstový faktor
Sources
1. Haas NB, Nathanson KL. Hereditary kidney cancer syndromes. Adv Chronic Kidney, DiS. 2014; 21(1): 81–90.
2. Leung C, Pan S, Shuch B. Management of renal cell carcinoma in young patients and patients with hereditary syndromes. Curr Opin Urol. 2016; 26(5): 396–404.
3. Huang KL, Mashl RJ, Wu Y, et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell. 2018; 173(2): 355–70.e14.
4. Shuch B, Vourganti S, Ricketts CJ, et al. Defining early‑onset kidney cancer: implications for germline and somatic mutation testing and clinical management. J Clin Oncol. 2014; 32(5): 431–437.
5. Menko FH, Maher ER. Diagnosis and Management of Hereditary Renal Cell Cancer. Recent Results Cancer Res. 2016; 205 : 85–104.
6. Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer 2015; 15(1): 55–64.
7. Bader HL, Hsu T. Systemic VHL gene functions and the VHL disease. FEBS Lett. 2012; 586(11): 1562–1569.
8. Richard S, Graff J, Lindau J, Resche F. Von Hippel‑Lindau disease. Lancet. 2004; 363(9416): 1231–1234.
9. Ürge T, Hora M, Toufarová P, et al. Renální karcinom u nemocných s morbus von Hippel-Lindau. Ces Urol 2007; 11(2): 93–97.
10. Nielsen SM, Rhodes L, Blanco I, et al. Von Hippel‑Lindau Disease: Genetics and Role of Genetic Counseling in a Multiple Neoplasia Syndrome. J Clin Oncol. 2016;34(18): 2172–2181.
11. Lattouf JB, Pautler SE, Reaume MN, et al. Structured assessment and followup for patients with hereditary kidney tumour syndromes. Can Urol Assoc J. 2016; 10(7–8):E214–E22.
12. Maddock IR, Moran A, Maher ER, et al. A genetic register for von Hippel‑Lindau disease. J Med Genet1996; 33(2): 120–127.
13. Ong KR, Woodward ER, Killick P, et al. Genotype‑phenotype correlations in von Hippel‑Lindau disease.Hum Mutat. 2007; 28(2): 143–149.
14. Deml KF, Schildhaus HU, Compérat E, et al. Clear cell papillary renal cell carcinoma and renal angiomyoadenomatous tumor: two variants of a morphologic,immunohistochemical, and genetic distinctentity of renal cell carcinoma. Am J Surg Pathol. 2015; 39(7): 889–901.
15. Maher ER, Neumann HP, Richard S. Von Hippel‑Lindau disease: a clinical and scientific review. Eur JHum Genet. 2011; 19(6): 617–623.
16. Walther MM, Choyke PL, Glenn G, et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999; 161(5): 1475–1479.
17. Duffey BG, Choyke PL, Glenn G, et al. The relationship between renal tumor size and metastases inpatients with von Hippel‑Lindau disease. J Urol. 2004; 172(1): 63–65.
18. Guo J, Tretiakova MS, Troxell ML, et al. Tuberous sclerosis‑associated renal cell carcinoma: a clinicopathologic study of 57 separate carcinomas in 18 patients. Am J Surg Pathol. 2014; 38(11): 1457–1467.
19. Vrtel R, Fillipová H, Vodicka R, et al. Tuberous sclerosis. Klin Onkol. 2009; 22(Suppl): S50–53.
20. Northrup H, Krueger DA, Group ITSCC. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. PediatrNeurol. 2013; 49(4): 243–254.
21. Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol. 2004; 19(9): 643–649.
22. Dixon BP, Hulbert JC, Bissler JJ. Tuberous sclerosis complex renal disease. Nephron Exp Nephrol. 2011;118(1): e15–20.
23. Tello R, Blickman JG, Buonomo C, Herrin J. Meta analysis of the relationship between tuberous sclerosis complex and renal cell carcinoma. Eur J Radiol. 1998; 27(2): 131–138.
24. Moch H, Ohashi R, Gandhi JS, Amin MB. Morphological clues to the appropriate recognition of hereditary renal neoplasms. Semin Diagn Pathol. 2018; 35(3): 184–192.
25. Palsgrove DN, Li Y, Pratilas CA, et al. Eosinophilic Solid and Cystic (ESC) Renal Cell Carcinomas HarborTSC Mutations: Molecular Analysis Supports an Expanding Clinicopathologic Spectrum. Am J Surg Pathol.2018; 42(9): 1166–1181.
26. Trpkov K, Hes O. New and emerging renal entities: a perspective post‑WHO 2016 classification. Histopathology 2019; 74(1): 31–59.
27. Trpkov K, Abou‑Ouf H, Hes O, et al. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC):Further Morphologic and Molecular Characterization of ESC RCC as a Distinct Entity. Am J Surg Pathol.2017; 41(10): 1299–308.
28. Carlo MI, Hakimi AA, Stewart GD, et al. Familial Kidney Cancer: Implications of New Syndromes and Molecular Insights. Eur Urol. 2019; 76(6): 754–764.
29. Dabora SL, Franz DN, Ashwal S, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF - D levels decrease. PLoS One. 2011; 6(9): e23379.
30. El‑Hashemite N, Walker V, Zhang H, Kwiatkowski DJ. Loss of Tsc1 or Tsc2 induces vascular endothelial growth factor production through mammalian target of rapamycin. Cancer Res. 2003; 63(17): 5173–5177.
31. Chen YB, Mirsadraei L, Jayakumaran G, et al. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma With Eosinophilic and Vacuolated Cytoplasm. Am J Surg Pathol. 2019; 43(1): 121–131.
32. Franz DN, Budde K, Kingswood JC, et al. Effect of everolimus on skin lesions in patients treated for subependymal giant cell astrocytoma and renal angiomyolipoma: final 4-year results from the randomized EXIST-1 and EXIST-2 studies. J Eur Acad Dermatol Venereol. 2018; 32(10): 1796–1803.
33. Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus long‑term use in patients with tuberous sclerosis complex: Four‑year update of the EXIST-2 study. PLoS One. 2017; 12(8): e0180939.
34. Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double‑blind, placebo‑controlled trial. Lancet 2013; 381(9869): 817–824.
35. Staehler M, Sauter M, Helck A, et al. Nephron‑sparing resection of angiomyolipoma after sirolimus pretreatment in patients with tuberous sclerosis. Int Urol Nephrol. 2012; 44(6): 1657–1661.
36. Ürge T, Pitra T, Chudáček Z, et al. Nové trendy v léčbě renalního angiomyolipomu. Ces Urol. 2015; 19(2): 106–117.
37. Moch H, Cubilla AL, Humphrey PA, Reuter VE, Ulbright TM. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs‑Part A: Renal, Penile, and Testicular Tumours. Eur Urol.2016; 70(1): 93–105.
38. Hora M, Ürge T, Kalusová K, et al. Novelizovaná klasifikace nádorů ledvin 2013 (International Society of Urological Pathology Vancouver Classification of Renal Neoplasia). Ces Urol 2014; 18(1): 9–20.
39. Schmidt LS, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma. Int J Nephrol Reno‑vasc, DiS. 2014; 7 : 253–260.
40. Trpkov K, Hes O, Agaimy A, et al. Fumarate Hydratase‑deficient Renal Cell Carcinoma Is Strongly Correlated With Fumarate Hydratase Mutation and Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome. Am J Surg Pathol. 2016; 40(7): 865–875.
41. Pivovarcikova K, Martinek P, Grossmann P, et al. Fumarate hydratase deficient renal cell carcinoma: Chromosomal numerical aberration analysis of 12 cases. Ann Diagn Pathol. 2019; 39 : 63–68.
42. Kuroda N, Ohe C, Kato I, et al. Review of hereditary leiomyomatosis renal cell carcinoma with focus on clinical and pathobiological aspects of renal tumors. Pol J Pathol. 2017; 68(4): 284–290.
43. Plevová P, Hladíková A, Tesařová M. Hereditary leiomyomatosis and renal cell cancer – HLRCC/multiple cutaneous and uterine leimomyomatosis – MCUL. Klin Onkol. 2012; 25 Suppl: S55–58.
44. Przybycin CG, Magi‑Galluzzi C, McKenney JK. Hereditary syndromes with associated renal neoplasia: a practical guide to histologic recognition in renal tumor resection specimens. Adv Anat Pathol. 2013; 20(4): 245–263.
45. Choyke PL, Walther MM, Glenn GM, et al. Imaging features of hereditary papillary renal cancers.J Comput Assist Tomogr. 1997; 21(5): 737–741.
46. Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/ VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013; 31(2): 181–186.
47. Schmidt LS, Nickerson ML, Angeloni D, et al. Early onset hereditary papillary renal carcinoma: germline missense mutations in the tyrosine kinase domain of the met proto‑oncogene. J Urol. 2004; 172(4 Pt1): 1256–1261.
48. Menko FH, van Steensel MA, Giraud S, et al. Birt‑Hogg‑Dubé syndrome: diagnosis and management.Lancet Oncol. 2009; 10(12): 1199–1206.
49. Hartman TR, Nicolas E, Klein‑Szanto A, et al. The role of the Birt‑Hogg‑Dubé protein in mTOR activation and renal tumorigenesis. Oncogene 2009; 28(13): 1594–1604.
50. Maher ER. Hereditary renal cell carcinoma syndromes: diagnosis, surveillance and management. WorldJ Urol. 2018; 36(12): 1891–1898.
51. Křepelová A, Puchmajerová A, Vasovčák P, Chocholatý M. Birt‑Hogg‑Dubé syndrome. Klin Onkol.2012; 25(Suppl): S18–20.
52. Pavlovich CP, Walther MM, Eyler RA, et al. Renal tumors in the Birt‑Hogg‑Dubé syndrome. Am J SurgPathol. 2002; 26(12): 1542–1552.
53. He H, Trpkov K, Martinek P, et al. „High‑grade oncocytic renal tumor“: morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch. 2018; 473(6): 725–738.
54. Stamatakis L, Metwalli AR, Middelton LA, Marston Linehan W. Diagnosis and management of BHD‑associated kidney cancer. Fam Cancer 2013; 12(3): 397–402.
55. Musil Z, Vícha A, Zelinka T, et al. Hereditary pheochromocytoma and paraganglioma. Klin Onkol.2012; 25 Suppl: S21–26.
56. Ricketts C, Woodward ER, Killick P, et al. Germline SDHB mutations and familial renal cell carcinoma.J Natl Cancer Inst. 2008; 100(17): 1260–1262.
57. Kuroda N, Yorita K, Nagasaki M, et al. Review of succinate dehydrogenase‑deficient renal cell carcinoma with focus on clinical and pathobiological aspects. Pol J Pathol. 2016; 67(1): 3–7.
58. Gill AJ, Hes O, Papathomas T, et al. Succinate dehydrogenase (SDH)-deficient renal carcinoma: a morphologically distinct entity: a clinicopathologic series of 36 tumors from 27 patients. Am J Surg Pathol.2014; 38(12): 1588–1602.
59. Rednam SP, Erez A, Druker H, et al. Von Hippel‑Lindau and Hereditary Pheochromocytoma/Paraganglioma Syndromes: Clinical Features, Genetics, and Surveillance Recommendations in Childhood. ClinCancer Res. 2017; 23(12): e68–e75.
60. Mester JL, Zhou M, Prescott N, Eng C. Papillary renal cell carcinoma is associated with PTEN hamartoma tumor syndrome. Urology. 2012; 79(5): 1187.e1–7.
61. Farley MN, Schmidt LS, Mester JL, et al. A novel germline mutation in BAP1 predisposes to familialclear‑cell renal cell carcinoma. Mol Cancer Res. 2013; 11(9): 1061–1071.
62. Popova T, Hebert L, Jacquemin V, et al. Germline BAP1 mutations predispose to renal cell carcinomas.Am J Hum Genet. 2013; 92(6): 974–980.
63. Chen JD, Morrison C, Zhang C, et al. Hyperparathyroidism‑jaw tumour syndrome. J Intern Med. 2003;253(6): 634–642.
64. Hora M, Hes O, Eret V, et al. Translokační renální karcinom Xp11.2 typu ASPL/TFE3. Ces Urol 2008;12(3): 186–193.
Labels
Paediatric urologist Nephrology UrologyArticle was published in
Czech Urology

2020 Issue 1
Most read in this issue
- Our experience with MRI/TRUS software fusion for targeted prostate biopsies
- Multi-resistant gram negative bacteria in urology
- Hereditary renal cell carcinoma syndromes
- Case report of a gigantic recurrent angiomyolipoma in a horseshoe kidney