
Vzácné dislipidemie, od fenotypu ke genotypu a léčbě: konsensus pracovní skupiny Evropské společnosti pro aterosklerózu
Autoři:
Robert; Hegele; Jan Borén; Henry N Ginsberg; Marcello Arca; Maurizio Averna; Christoph J Binder; Laura Calabresi; M. John Chapman; Marina Cuchel; Arnold Von Eckardstein; Ruth Frikke-Schmidt; Daniel Gaudet; G. Kees Hovingh; Florian Kronenberg; Dieter Lütjohann; Klaus G Parhofer; Frederick J Raal; Kausik K Ray; Alan T Remaley; Jane K Stock; Erik S Stroes; Lale Tokgözo Lu; Alberico L Catapano
Působiště autorů:
Department of Vascular Medicine, Academic Medical Center, Amsterdam, Netherlands (Prof G K Hovingh MD, Prof E S Stroes MD)
; Department of Molecular and Clinical Medicine, University of Gothenburg and Sahlgrenska University Hospital, Gothenburg, Sweden (Prof J Borén MD)
; and Lipid Clinic, Chicoutimi Hospital, Chicoutimi, QC, Canada (Prof D Gaudet)
; European Atherosclerosis Society, Gothenburg, Sweden (J K Stock PhD)
; Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, Milan, Italy (Prof A L Catapano PhD)
; Medizinische Klinik IV-Grosshadern, University of Munich, Munich, Germany (Prof K G Parhofer MD)
; Imperial Centre for Cardiovascular Disease Prevention, Department of Primary Care and Public Health, Imperial College London, London, UK (Prof K K Ray MD)
; National Institute for Health and Medical Research (INSERM), Sorbonne University and Pitié-Salpétrière University Hospital, Paris, France (Prof M J Chapman DSc)
; Division of Translational Medicine and Human Genetics, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA (M Cuchel MD)
; Department of Laboratory Medicine, Medical University of Vienna, Vienna, Austria (Prof C J Binder MD)
; Centro Grossi Paoletti, Dipartimento di Scienze Farmacologichee Biomolecolari, Università degli Studi di Milano, Milan, Italy (Prof L Calabresi PhD)
; Department of Health Promotion Sciences Maternal and Infantile Care, Internal Medicine and Medical Specialities, University of Palermo, Palermo, Italy (Prof M Averna MD)
; Department of Internal Medicine and Allied Sciences, Center for Rare Disorders of Lipid Metabolism, Sapienza University of Rome, Rome, Italy (M Arca MD)
; Department of Medicine, Vagelos College of Physicians and Surgeons, Columbia University, New York, NY, USA (Prof H N Ginsberg MD)
; Division of Genetic Epidemiology, Department of Medical Genetics, Molecular and Clinical Pharmacology, Medical University of Innsbruck, Innsbruck, Austria (Prof F Kronenberg MD)
; Institute of Clinical Chemistry, University Hospital Zurich, Zurich, Switzerland (Prof A von Eckardstein MD)
; Institute of Clinical Chemistry and Clinical Pharmacology, University Hospital Bonn, Bonn, Germany (D Lütjohann PhD)
; Department of Clinical Medicine, Faculty of Health and Medical Science, University of Copenhagen, Copenhagen, Denmark (Prof R Frikke-Schmidt MD)
; Carbohydrate and Lipid Metabolism Research Unit, Division of Endocrinology and Metabolism, Department of Medicine, Faculty of Health Sciences, University of the Witwatersrand, Parktown, Johannesburg, South Africa (Prof F J Raal MD)
; Department of Clinical Biochemistry, Rigshospitalet Copenhagen University Hospital, Copenhagen, Denmark (Prof R Frikke-Schmidt)
; Lipoprotein Metabolism Section, Translational Vascular Medicine Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD, USA (Prof A T Remaley MD)
; Clinical Lipidology and Rare Lipid Disorders Unit, Community Genomic Medicine Center, Department of Medicine, Université de Montréal, Montreal, QC, Canada (Prof D Gaudet MD)
; Department of Cardiology, Hacettepe University Faculty of Medicine, Ankara, Turkey (Prof L Tokgözoğlu MD)
; ECOGENE, Clinical and Translational Research Center (Prof D Gaudet)
; and IRCCS MultiMedica, Milan, Italy (Prof A L Catapano)
; Department of Medicine and Robarts Research Institute, Schulich School of Medicine and Dentistry, Western University, London, ON, Canada (Prof R A Hegele MD)
Vyšlo v časopise:
AtheroRev 2021; 6(Supplementum 1): 3-18
Kategorie:
Doporučené postupy
Souhrn
Sekvenování genomu a genové terapie jsou slibnými strategiemi, které by měly zkvalitnit péči o pacienty se vzácnými poruchami lipoproteinů a souvisejícími dyslipidemiemi. V praxi je však běžné „poddiagnostikování“ a nedostatečná léčba zejména z důvodu významné interindividuální variability v genetické příčině i výsledném fenotypu. Evropská společnost pro aterosklerózu tedy jmenovala pracovní skupinu, aby vypracovala praktická klinická doporučení zaměřená na pacienty s extrémními (nízkými nebo vysokými) plazmatickými koncentracemi cholesterolu v lipoproteinech o nízké hustotě, triglyceridů nebo cholesterolu v lipoproteinech o vysoké hustotě. Tato doporučení rovněž reflektují nedostatek kvalitních dat o prevalenci a důsledcích těchto onemocnění. Pro zlepšení opatření v péči o pacienty se vzácnými dyslipidemiemi jsou nutné rozsáhlejší registry.
Úvod
Co je vzácné onemocnění? Univerzální definice je obtížná, avšak průměrně se celková prevalence vzácného onemocnění odhaduje na 40–50 případů na 100 000 lidí podle rozdílů v definici v jednotlivých zemích [1]. Kritéria používaná regulačními úřady v Evropě a USA jsou rovněž v souladu s tímto odhadem (tab. 1) [2,3].

Ačkoliv jednotlivé vzácné onemocnění postihuje nízký počet lidí, všechna vzácná onemocnění společně představují značnou zátěž pro zdravotnictví. Vzhledem k více než 7 000 dodnes identifikovaných vzácných onemocnění je postižen přibližně jeden z 12 lidí neboli přibližně 36 miliónů lidí v Evropě (a kumulativně přibližně 500 miliónů lidí na světě) [4]. Správný postup léčby pro snížení této zátěže je tedy naprosto zásadní pro kliniky, plátce i politiky. Pacienti a jejich rodiny často podstupují dlouhý diagnostický proces, než se zjistí správná diagnóza [5]. Protože více než 80 % vzácných onemocnění má genetickou příčinu, má vyšetření genomu naprosto zásadní roli v diagnostice a léčbě i při vývoji nových terapií.
Pokroky v oblasti vzácných dyslipidemií společně s klesající cenou za sekvenování genomu a bioinformatiku umožňují použít při léčbě těchto pacientů přístupy personalizované medicíny. Klinická praxe však často zůstává pozadu. Toto zpoždění lze vysvětlit několika faktory, jako jsou nedostatek kvalitních informací o prevalenci těchto onemocnění, interindividuální variabilita fenotypu a nejistota ohledně relativní důležitosti fenotypu versus genotypu při poskytování zdravotní péče. Další překážkou pro stanovení diagnózy je skutečnost, že genetické varianty s malým efektem mohou společně ovlivnit vyjádření fenotypu v rámci polygenního působení [6]. Všechny tyto faktory komplikují diagnostiku, léčbu a přístup k léčbě vzácných dyslipidemií.
Cílem tohoto konsensu pracovní skupiny Evropské společnosti pro aterosklerózu (EAS) je vyřešit tyto nejistoty poskytnutím vysvětlení základní patofyziologie a praktického klinického doporučení pro vzácné dyslipidemie spojené s extrémními hladinami (nízkými i vysokými) cholesterolu v lipoproteinech o nízké hustotě (LDL), triglyceridů, nebo cholesterolu v lipoproteinech o vysoké hustotě (HDL). Ačkoli má genetické testování v definitivní diagnóze zásadní roli, klinický postup určuje převážně vyjádřený fenotyp.
Přehled vzácných poruch lipoproteinů
Jako extrémní biochemická odchylka s, nebo bez tělesných příznaků, je definováno nejméně 25 monogenních dyslipidemií, které obvykle odpovídají vzorci autosomálně dominantní, kodominantní, nebo recesivní dědičnosti [7]. Tyto poruchy jsou způsobeny vzácnými mutacemi postihujícími celkem 23 genů (tab. 2), na které cílí diagnostická sekvenace DNA (tzv. pan-dyslipidemické panely) [7] a podle nichž jsou definovány bioinformatické parametry pro stanovení profilu variant z výsledků sekvenování celého exomu nebo genomu. Mutace různých genů mohou příležitostně způsobit stejný fenotyp (např. dominantní formu familiární hypercholesterolemie), a naopak protichůdné mutace stejného genu (ztráta funkce versus zvýšení aktivity) mohou způsobit opačné fenotypy (např. mutace APOB a PCSK9 způsobující buď vysoké, nebo nízké koncentrace LDL-cholesterolu). Obr. 1 schematicky zobrazuje metabolizmus lipoproteinů zacílený na produkty genů, které způsobují monogenní dyslipidemie.
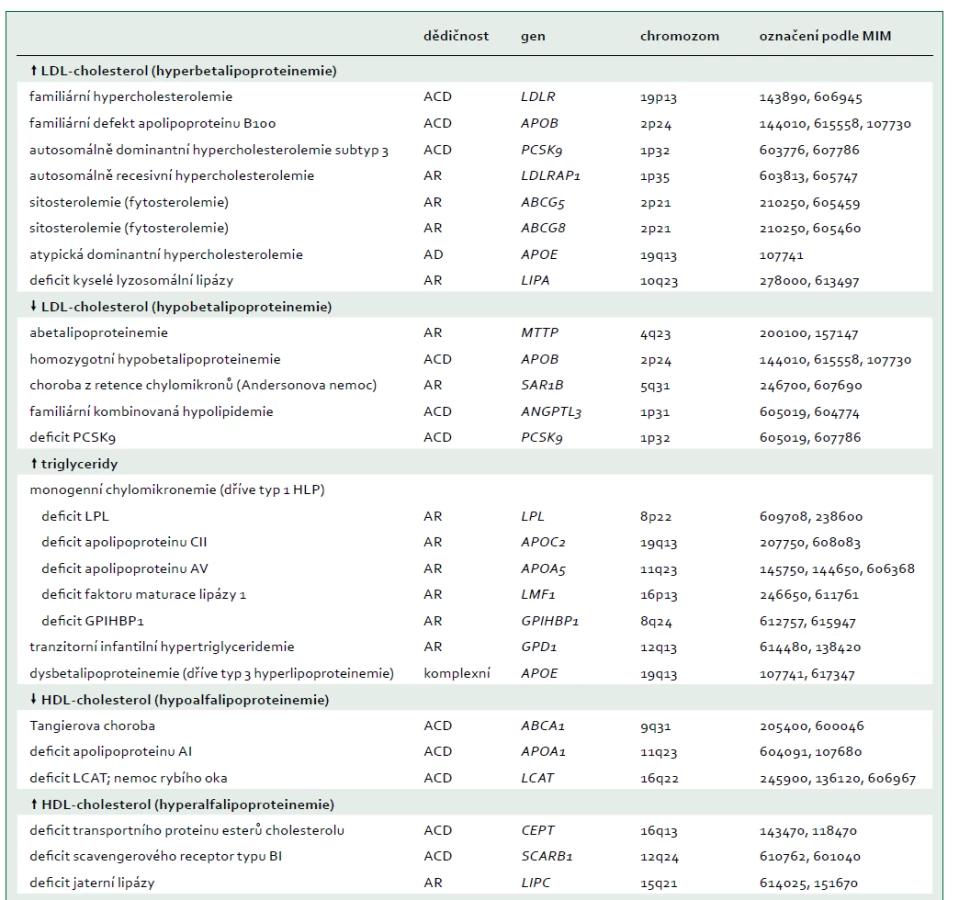
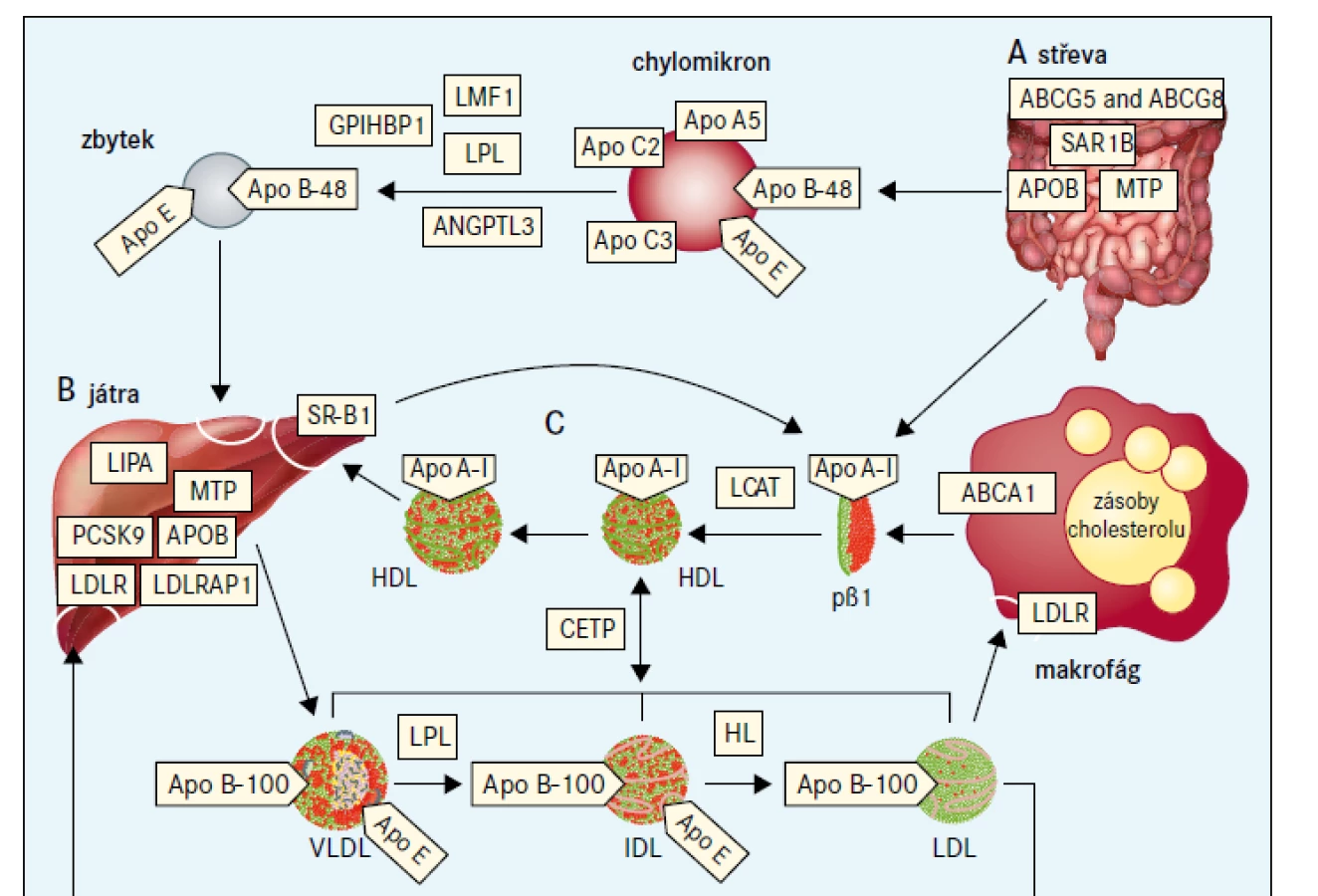
ABCA1 – transportní protein rodiny ABC (ATP-binding cassette) typ A1 ABCG5 – transportní protein rodiny ABC (ATP-binding cassette) typ 5 ABCG8 – transportní protein rodiny ABC (ATP-binding cassette) typ 8 ANGPTL3 – protein 3 podobný angiopoietinu Apo – apolipoprotein CETP – transportní protein esterů cholesterolu GPIHBP1 – protein 1 vázající HDL ukotvený na glykosylfosfatidylinositolu HL – jaterní lipáza
IDL – lipoprotein se střední hustotou LCAT – lecitin cholesterol acyl transferáza LDLR – jaterní receptor LDL LDLRAP1 – LDL receptor-adaptorový protein LIPA – lyzosomální kyselá lipáza LMF1 – faktor maturace lipázy 1 LPL – lipoproteinová lipáza MTP – mikrosomální triglyceridový transportní protein pβ1 – prebeta1-HDL PCSK9 – proprotein konvertáza subtilizin/kexin typu 9 SAR1B – SAR1 homolog B GTPázy SR-BI – scavengerový receptor
Poruchy spojené s LDL
Lipoproteiny obsahující apolipoprotein B zahrnují LDL, lipoproteiny se střední hustotou [IDL], včetně těch, které odpovídají zbytkům lipoproteinů s velmi nízkou hustotou [VLDL], VLDL, chylomikrony a jejich zbytkové částice a lipoprotein (a). Všechny jsou proaterogenní a mají klíčovou roli při transportu cholesterolu a triglyceridů v cirkulaci [8]. Tyto částice byly dříve označované podle elektroforetické pohyblivosti jako třída beta a pre-beta.
Poruchy charakterizované velmi vysokou hladinou LDL-cholesterolu
Patofyziologie
Hyperbetalipoproteinemie je určujícím znakem pro několik vzácných dyslipidemií se značně zvýšenou hladinou LDL-cholesterolu nebo apolipoproteinu B100. Tyto stavy vyplývají převážně z narušení interakce mezi LDL-částicí a LDL-receptorem. Výslednou klinickou poruchou je familiární hypercholesterolemie, při které je zásadní vadou zpožděná clearance LDL z plazmy, což vede k hypercholesterolemii, tělesným příznakům (včetně arcus cornealis, xantelazmat a šlachových xantomů; obr. 2) a, pokud se neléčí, předčasnému aterosklerotickému kardiovaskulárnímu onemocnění [9].
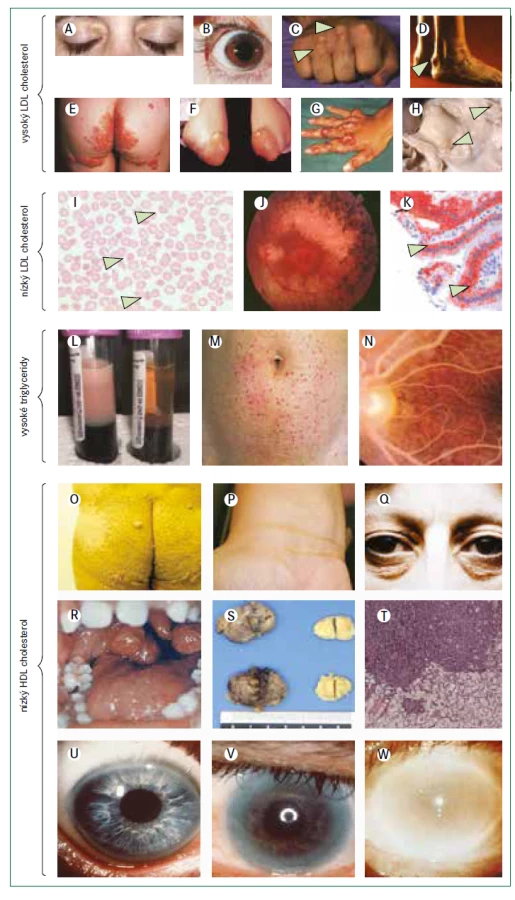
Obrázky B, E–H a M jsou reprodukovány z Davignon a Dufour [101] se svolením Clinical Publishing. Obrázky B, C a D jsou reprodukovány z Genest et al se svolením Elsevier [102]. Obrázek J je reprodukován z Hamel [103]. Obrázek K je reprodukován ze Sassolas et al [104]. Obrázky L a N jsou reprodukovány z Yuan et al se svolením CMAJ [105]. Obrázky O a P jsou reprodukovány ze Santos et al [79] se svolením Elsevier. Obrázky S a T jsou reprodukovány z Ravesloot et al [106] se svolením Elsevier. Všechny ostatní obrázky jsou z vlastní sbírky autorů.
O diagnostice a léčbě familiární hypercholesterolemie již byla vypracována řada komplexních přehledových článků [9,10]. Heterozygotní familiární hypercholesterolemie je nejčastější dědičná metabolická porucha, která způsobuje aterosklerotické kardiovaskulární onemocnění, a postihuje asi 1 z 200–250 lidí [9,10]. Protože heterozygotní familiární hypercholesterolemie není vzácná porucha (podle evropských i americkými definic tohoto pojmu), nebudeme se jí v tomto konsensu podrobně věnovat. Naproti tomu homozygotní familiární hypercholesterolemie je velmi vzácné onemocnění, které postihuje přibližně 1 ze 160 000–300 000 lidí na celém světě [11].
Familiární hypercholesterolemie je autosomálně dominantní porucha. Většina jedinců s geneticky potvrzenou heterozygotní familiární hypercholesterolemií a jedinců s homozygotní familiární hypercholesterolemií má 1, respektive 2 mutované alely genu LDLR, což vede k defektní nebo nulové funkci LDL-receptoru. Heterozygotní mutace v jiných genech, včetně APOB a PCSK9, vysvětlují méně než 10 % případů heterozygotní familiární hypercholesterolemie a 2 mutované alely těchto genů a LDLRAP1 (také nazývané autosomálně recesivní hypercholesterolemie – ARH) způsobují fenotyp, který se podobá homozygotní familiární hypercholesterolemii [11,12].
V genu LDLR bylo identifikováno více než 2 300 jedinečných mutací způsobujících familiární hypercholesterolemii [13]. Z mutací APOB je nejčastěji pozorována Arg3527Gln (záměna argininu na glutamin na pozici 3527), která narušuje interakci apolipoproteinu B s LDL-receptorem [14]. Asi 50 dalších pravděpodobně patogenních mutací APOB vede k hyperlipidemii [13]. Mnohé z těchto mutací postihují arginin v doméně, která se váže na receptor a je kódována především exonem 26 [14]. U pacientů s familiární hypercholesterolemií bylo hlášeno více než 30 mutací v genu kódujícím proprotein konvertázu subtilizin/kexin typu 9 (PCSK9) způsobujících zvýšení funkce tohoto enzymu; dohromady však tyto mutace představují méně než 1 % všech případů familiární hypercholesterolemie [15]. Dalším potenciálně kauzálním genem byl STAP1. Dokud se však neobjeví přesvědčivější data, nebudou velmi vzácné mutace v tomto genu považovány za příčinu familiární hypercholesterolemie. V poslední řadě, nejméně 20 % pacientů odeslaných do specializované péče lipidologa se suspektní heterozygotní familiární hypercholesterolemií nese polygenní predispozici k vysokým hladinám LDL-cholesterolu [16]. Podrobná diskuse o polygenní etiologii dyslipidemií je nad rámec tohoto konsensu; na toto téma jsou k dispozici jiné práce [6,17].
Klinický obraz a diagnóza
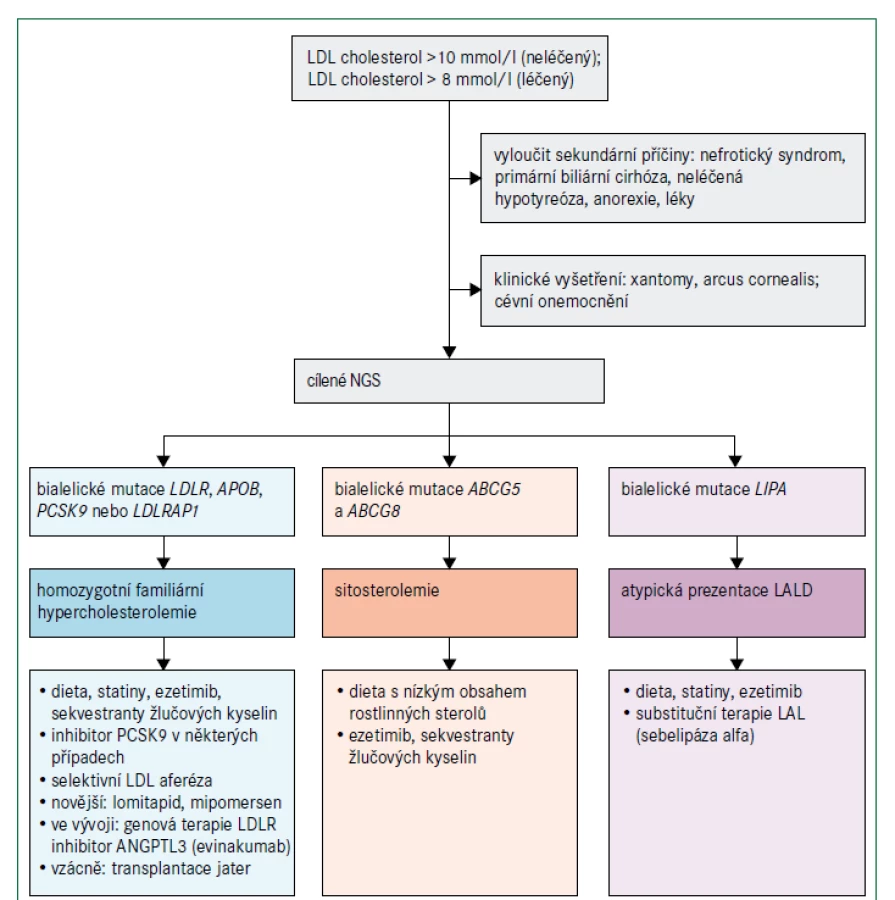
Vzhledem k poslání tohoto konsensu se zaměříme na pacienty s extrémně zvýšenými hladinami LDL-cholesterolu, zvláště na homozygotní familiární hypercholesterolemii (obr. 3), která je velmi vzácná. Historicky hladina LDL-cholesterolu vyšší než 8 mmol/l (> 300 mg/dl) při léčbě nebo hladina LDL-cholesterolu vyšší než 10 mmol/l (> 400 mg/dl) bez léčby spolu se zřejmými kožními nebo šlachovými xantomy před dosažením věku 10 let byly dostatečným důvodem pro stanovení diagnózy homozygotní familiární hypercholesterolemie. Nyní víme, že klinický obraz se může lišit zejména z důvodu genetické heterogenity familiární hypercholesterolemie [11]. Diagnóza se stále primárně opírá o klinické vyšetření; užitečné jsou bodovací systémy stejně jako cílené sekvenování DNA, jestliže prokáže bialelické patogenní mutace v příslušných genech. Pacienti s homozygotní familiární hypercholesterolemií mají nejčastěji patogenní mutace v genu LDLR, obvykle 2 různé mutace (složený heterozygot) nebo, vzácněji, stejnou mutaci (jednoduchý nebo pravý homozygot) [11,17]. Závažnost zvýšení plazmatického LDL-cholesterolu a klinické projevy závisí nejen na genu, který je příčinnou tohoto stavu, ale i na typu mutace, ačkoliv existuje i značná interindividuální variabilita [11,17]. Protože pro riziko aterosklerotického kardiovaskulárního onemocnění je klíčovým faktorem individuální hladina LDL-cholesterolu, a nikoliv typ mutace, měla by se podle ní řídit i intenzita léčby [11].
Jiné vzácné dyslipidemie mohou mít klinický obraz podobný homozygotní familiární hypercholesterolemii, i když obvykle s nižšími hladinami LDL-cholesterolu (obr. 3). β-sitosterolemie (fytosterolemie), autosomální recesivní porucha způsobená mutacemi v genech ABCG5 a ABCG8, které kódují transportní proteiny rodiny ABC (ATP binding cassette) podrodiny G, typu 5, respektive 8, vede k retenci necholesterolových sterolů a je charakterizována atypickou xantomatózou se zvýšenou koncentrací rostlinných sterolů a stanolů (fytosterolů) s i bez zvýšené hladiny LDL-cholesterolu a s proměnlivou náchylností k časnému aterosklerotickému kardiovaskulárnímu onemocnění (obr. 2). Velmi příležitostně jsou některé mírnější případy zvýšeného LDL-cholesterolu se současnou hepatosplenomegalií a s proměnlivými koncentracemi triglyceridů výsledkem deficitu lyzosomální kyselé lipázy (nazývaného také onemocnění z ukládání esterů cholesterolu nebo u pediatrických pacientů Wolmanova choroba), což je autosomálně recesivní poškození genu LIPA [18]. Definitivní diagnóza se u těchto vzácných poruch stanoví pomocí sekvenování DNA.
Současná a budoucí léčba
Léčba homozygotní familiární hypercholesterolemie vychází z algoritmů pro heterozygotní familiární hypercholesterolemii, které jsou dobře zavedené a obvykle kromě diety a úpravy životního stylu zahrnují také kombinaci maximálně tolerovaného statinu, ezetimibu a inhibitoru PCSK9 [9–11]. U homozygotní familiární hypercholesterolemie je navíc zásadní aferéza lipoproteinů vzhledem k závažnosti zvýšení LDL-cholesterolu, velkému riziku aterosklerózy a refrakternosti vůči jiným způsobům léčby [11,19]. Volbu léčebné strategie u homozygotní familiární hypercholesterolemie může ovlivnit výsledek genetického testování, protože monoklonální protilátka proti PCSK9 evolokumab je neúčinná u pacientů se 2 nulovými mutacemi LDLR, ale může být účinná v přítomnosti mutací LDLR s částečně zachovanou funkcí receptoru. Protilátky proti PCSK9 jsou účinné, jsou-li přítomny bialelické mutace PCSK9 způsobující zvýšení funkce proteinu (obr. 3) [20]. Další doplňkovou terapeutickou možností u pacientů s homozygotní familiární hypercholesterolemií je perorální inhibitor mikrosomálního triglyceridového transportního proteinu lomitapid [21,22]. Účinnost této léčby je maximalizována dodržováním nízkotučné stravy (< 20 % energie získané z tuku) s dávkováním mimo dobu jídla, aby se minimalizovaly gastrointestinální příznaky; v důsledku mechanizmu účinku se však může objevit jaterní steatóza. Mipomersen, antisense oligonukleotid apolipoproteinu B 2. generace, byl k dispozici v USA až do května 2018, kdy byl prodej přerušen pro obavy o bezpečnost pro zvýšení jaterních transamináz a jaterní steatózu. V Evropě mipomersen schválen nebyl. Evinakumab (monoklonální protilátka proti ANGPTL3 ) a genová terapie LDLR by mohly mít terapeutický potenciál jako doplňková léčba [22,23]. Ojediněle lze u pacientů s homozygotní familiární hypercholesterolemií zvážit transplantaci jater [11]. Pokud je diagnostikována sitosterolemie, léčba se významně liší: aferéza není nutná a hyperlipidemie často dobře reaguje na snížení příjmu sterolu v potravě a na léčbu ezetimibem nebo sekvestranty žlučových kyselin [24]. Pokud je diagnostikována deficience lyzosomální kyselé lipázy, léčba zahrnuje substituci enzymu infuzí sebelipázy alfa [25].
Léčba lidí s homozygotní familiární hypercholesterolemií si zaslouží zvážení celé řady dalších okolností souvisejících s genetickým poradenstvím, kaskádovým screeningem k identifikaci rodinných příslušníků postižených heterozygotní familiární hypercholesterolemií a u žen opatření týkající se antikoncepce a těhotenství. Další informace jsou k dispozici v jiných přehledových článcích [9–11].
Poruchy charakterizované velmi nízkou hladinou LDL-cholesterolu
Jako primární hypobetalipoproteinemie se označuje skupina dyslipidemií charakterizovaných velmi nízkou nebo nulovou koncentrací LDL-cholesterolu a apolipoproteinu B v plazmě. Další lipidy a lipoproteiny mohou být také změněny podle genu a závažnosti mutace, nebo mutací (tab. 2), které jsou příčinou [26,27].
Patofyziologie
Hypobetalipoproteinemie může vzniknout v důsledku snížené produkce nebo zvýšeného katabolizmu lipoproteinů obsahujících apolipoprotein B. Mutace se ztrátou funkce v genu MTTP, kódujícím mikrosomální triglyceridový transportní protein (MTP), způsobují abetalipoproteinemii (nazývanou také Bassenův–Kornzweigův syndrom), autosomálně recesivní poruchu, charakterizovanou absencí produkce VLDL a chylomikronů, což vede k neměřitelným plazmatickým hladinám LDL-cholesterolu a apolipoproteinu B a velmi nízké hladině triglyceridů a celkového cholesterolu (< 0,33 mmol/l) [28]. Dosud bylo popsáno více než 30 různých mutací spojených se ztrátou funkce v genu MTTP, z nichž všechny zásadně narušují schopnost lipidů vázat se do nově vznikajících lipoproteinů obsahujících apolipoprotein B [28].
Homozygotní familiární hypobetalipoproteinemie (FHBL) klinicky připomíná abetalipoproteinemii. FHBL je autosomální kodominantní porucha zahrnující gen APOB a je charakterizována velmi nízkými koncentracemi apolipoproteinu B (nižšími než 5. percentil pro věk a pohlaví) a LDL-cholesterolu (obvykle < 1,0 mmol/l) [29]. Na rozdíl od mutací ovlivňujících vazbu na LDL-receptor, které způsobují opačný fenotyp (tj. familiární hypercholesterolemii), mutace v APOB způsobující FHBL narušují integritu lipoproteinových částic. Více než 60 různých patogenních mutací v APOB mimo doménu vázající se k receptoru je spojeno se strukturními defekty proteinu, často se sekrecí zkrácených forem apolipoproteinu B (např. apolipoprotein B9 [což odpovídá 9 % celé délky proteinu] až apolipoprotein B89 [což odpovídá 89 % celé délky proteinu]), sníženou sekrecí VLDL a zvýšeným katabolizmem VLDL a LDL, což vede ke snížení koncentrací cholesterolu a triglyceridů v oběhu [30–32]. Jinou příčinou primární hypobetalipoproteinemie mohou být mutace spojené se ztrátou funkce v SAR1B, genu kódujícím Sar1 homolog B GTPázy, v ANGPTL3, genu kódujícím protein 3 podobný angiopoietinu, a PCSK9. Bialelické mutace v SAR1B způsobují autosomálně recesivní chorobu z retence chylomikronů (známá také jako Andersonova choroba), která se projevuje selháním sekrece chylomikronů z enterocytů, ačkoliv přesná příčina přesně známa není (tab. 2) [34,35]. V poslední řadě více než 30 různých mutací spojených se ztrátou funkce v PCSK9 vede ke snížení lyzosomální degradace LDL-receptoru a jeho zvýšené recyklaci na buněčný povrch, což vede ke zvýšenému katabolizmu částic LDL, čímž se snižuje koncentrace LDL-cholesterolu v plazmě [15].
Klinický obraz a diagnóza
Obr. 4 popisuje algoritmus pro diagnostiku a léčebný postup poruch charakterizovaných velmi nízkými koncentracemi LDL-cholesterolu. Abetalipoproteinemie a homozygotní FHBL jsou spojeny s nedetekovatelnými koncentracemi LDL-cholesterolu a apolipoproteinu B při přímém vyšetření, koncentrace triglyceridů jsou velmi nízké a téměř veškerý cholesterol v plazmě je transportován částicemi HDL. Protože exogenní vitaminy rozpustné v tucích jsou absorbovány prostřednictvím chylomikronů a transportovány prostřednictvím lipoproteinů obsahujících apolipoprotein B, poruchy abetalipoproteinemie, homozygotní FHBL a choroba z retence chylomikronů vedou k závažným deficiencím vitaminů rozpustných v tucích. Klinické projevy (obr. 2) zahrnují akantocytózu s mírnou anémií od narození, malabsorpci tuků a poruchu růstu v raném dětství; pozdější projevy deficitu vitaminů rozpustných v tucích jsou šeroslepost, atypická retinitis pigmentosa, osteomalacie nebo křivice, příznaky zadních provazců, spinocerebelární ataxie, periferní neuropatie a prodloužený protrombinový čas (nebo INR – mezinárodní normalizovaný poměr/International Normalised Ratio) [28,29,33]. Obě onemocnění lze odlišit podle toho, že heterozygotní rodiče pacientů s homozygotní FHBL mají snížené hladiny LDL-cholesterolu, zatímco rodiče pacientů s abetalipoproteinemií mají normální lipidové profily. Pacienti s heterozygotní hypobetalipoproteinemií mají také zvýšené riziko jaterní steatózy, ale současně snížené riziko aterosklerotického kardiovaskulárního onemocnění [28].
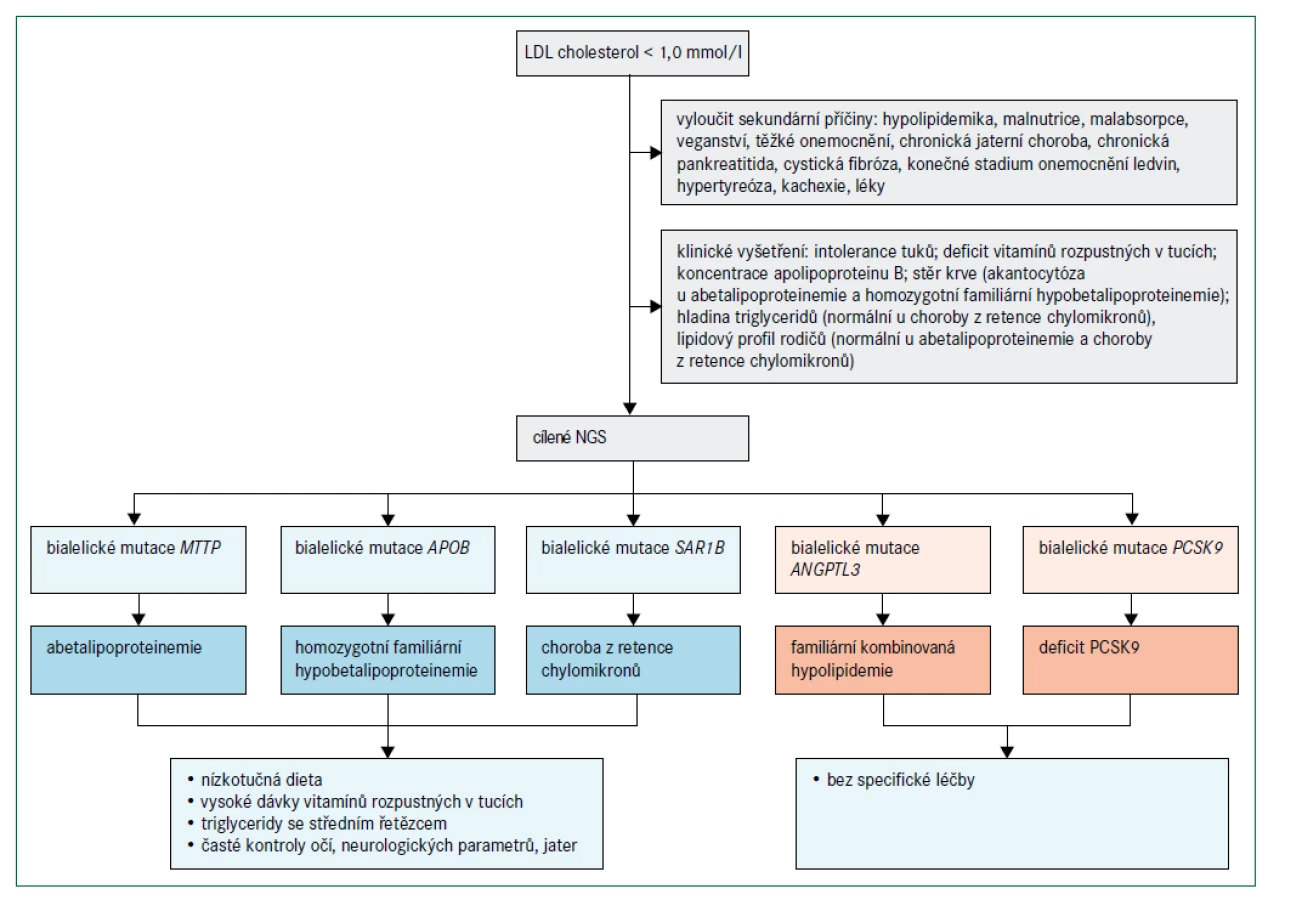
O chorobě z retence chylomikronů lze uvažovat, jestliže kojenec neprospívá a přítomná je i těžká malabsorpce se steatoreou a nedostatkem vitaminů rozpustných v tucích [33]. Choroba z retence chylomikronů se projevuje relativně normálními hladinami triglyceridů, absencí apolipoproteinu B48 a chylomikronů po tukové zátěži a méně závažným postižením očí než při abetalipoproteinemii. Heterozygotní rodiče dětí s chorobou z retence chylomikronů mají normální lipidové profily. Naopak heterozygoti pro deficit ANGPTL3 mají hladinu celkového cholesterolu, LDL-cholesterolu a triglyceridů přibližně o 50 % nižší, než je norma, a relativně normální hodnotu HDL-cholesterolu, zatímco homozygoti mají velmi výrazně snížené hladiny celkového cholesterolu, LDL-cholesterolu, triglyceridů i HDL-cholesterolu, ale chybí zde deficience vitaminů nebo jiné specifické klinické projevy, a naopak je pravděpodobně přítomný protektivní účinek před aterosklerotickým kardiovaskulárním onemocněním [34,35]. Jedinci s bialelickými mutacemi spojenými se ztrátou funkce PCSK9 mají méně snížené koncentrace LDL-cholesterolu než u abetalipoproteinemie, homozygotní FHBL nebo choroby z retence chylomikronů a nemají žádné patologické fenotypové projevy [36]. Diagnóza je potvrzena, pokud jsou funkčně významné mutace detekovány sekvenováním DNA [15]. Při vyšetření pacientů s hypobetalipoproteinemií je nutné vyloučit sekundární příčiny (např. chronické onemocnění jater, chronickou pankreatitidu, cystickou fibrózu, onemocnění ledvin v konečném stadiu, hypertyreózu, kachexii a malabsorpci), obr. 4 [26,27].
Současná léčba
Včasná diagnóza a léčba jsou nezbytné k prevenci dlouhodobých oftalmologických a neurologických komplikací u pacientů s abetalipoproteinemií, homozygotní FHBL a chorobou z retence chylomikronů. Mezi obecné principy léčby těchto tří onemocnění patří dieta s omezením tuků (s triglyceridy se středním řetězcem nebo bez nich), suplementace esenciálních mastných kyselin a vysoké perorální dávky vitaminů A, D, E a K, což do značné míry umožní korigovat deficience pravděpodobně prostřednictvím vstřebávání triglyceridů se středním řetězcem přes portální žílu [26–29]. U nositele bialelických mutací spojených se ztrátou funkce v PCSK9 a ANGPTL3 není vyžadována žádná zvláštní léčba. Heterozygotní příbuzní 1. stupně mají buď normální lipidové profily (u mutací genů MTTP a SAR1B), nebo mírnou až střední hypolipidemii (u mutací genů APOB, PCSK9 a ANGPTL3 ). U pacientů s heterozygotními mutacemi spojenými se ztrátou funkce v APOB se může objevit jaterní steatóza [26–29,37]. Ačkoliv klinické následky a terapie této komplikace nebyly dosud určeny, lze doporučit suplementaci vitaminů rozpustných v tucích pro korekci případných deficiencí. Hypobetalipoproteinemie spojená s mutacemi se ztrátou funkce v ANGPTL3 nebo PCSK9 je naopak zcela benigní, nebo má dokonce i protektivní účinek a nevyžaduje žádnou specifickou léčbu.
Syndromy s chylomikronemií
Hypertriglyceridemie je definována jako koncentrace triglyceridů nalačno vyšší než 2,0 mmol/l (ačkoli někdy je za prahovou hodnotu považována hladina > 1,7 mmol/l). Závažná hypertriglyceridemie, definovaná jako koncentrace triglyceridů vyšší než 10 mmol/l, postihuje 0,1–0,2 % populace. Hladiny triglyceridů nalačno zvýšené na tuto úroveň téměř vždy znamenají patologickou přítomnost chylomikronů [38]. Většina jedinců se zjištěnými genetickými příčinami této odchylky má polygenní predispozici definovanou jako akumulace běžných variant s malým individuálním účinkem na koncentraci triglyceridů, nebo heterozygotní vzácnou mutaci se ztrátou funkce s neúplnou penetrancí (nebo více mutací) [39]. Nanejvýš 1–2 % dospělých se závažnou hypertriglyceridemií má monogenní příčinu definovanou jako recesivní (bialelická) vzácná varianta s významným účinkem (tj. buď jednoduchý homozygot, nebo složený heterozygot) v genech podílejících se na regulaci metabolizmu lipoproteinů bohatých na triglyceridy [39]. Široce používaný termín syndrom familiární chylomikronemie je synonymum pro námi preferovaný termín monogenní chylomikronemie. Ve srovnání s pacienty, kteří mají mnohem častější multifaktoriální nebo polygenní chylomikronemii, lze u pacientů s monogenní chylomikronemií pozorovat následující: hypertriglyceridemie se objevuje v mladším věku, včetně dětství; menší pravděpodobnost obezity a spíše bez sekundárních faktorů, koncentrace triglyceridů nalačno může překročit 20 mmol/l; vyšší celoživotní riziko rozvoje akutní pankreatitidy (tj. 60–70 % v porovnání s 5–10 % u multifaktoriální chylomikronemie); mnohem nižší koncentrace apolipoproteinu B100 a značná rezistence vůči lékům snižujícím triglyceridy.
Patofyziologie
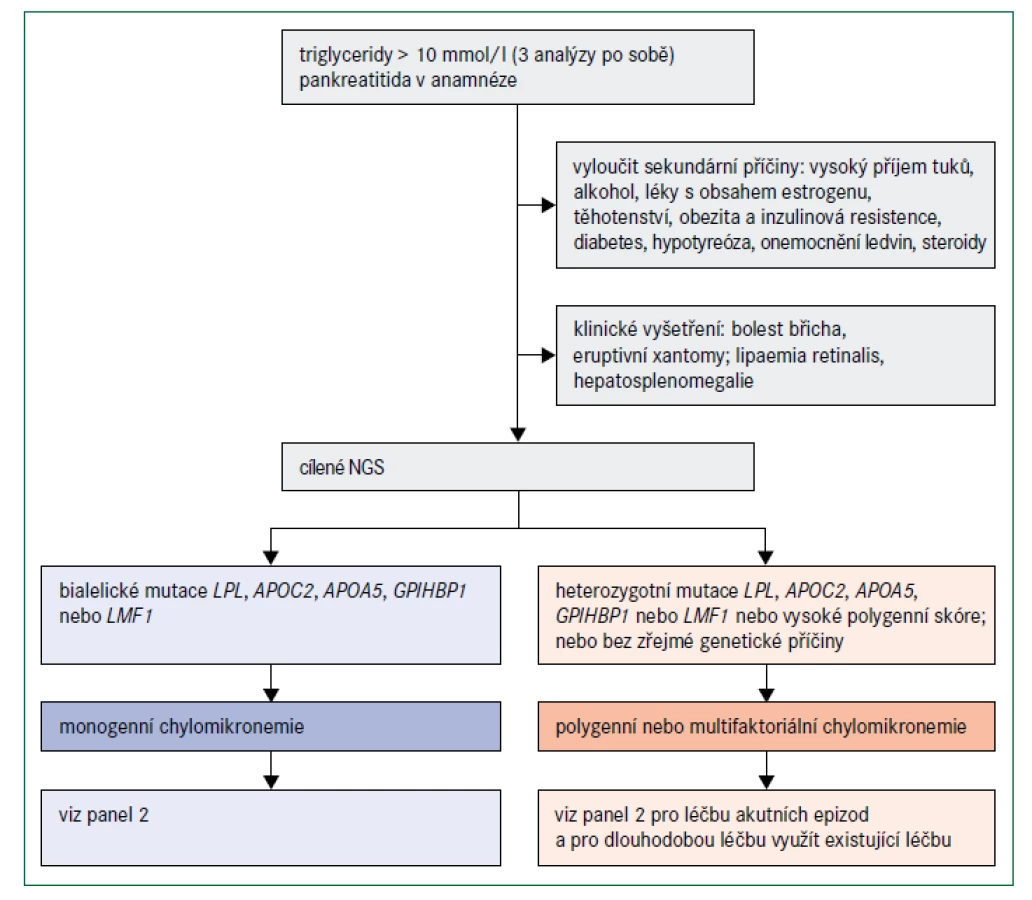
Zatímco nadměrná produkce VLDL játry je nejčastější příčinou mírné až střední hypertriglyceridemie, monogenní závažná hypertriglyceridemie je důsledkem silně nebo zcela narušené lipolýzy lipoproteinů bohatých na triglyceridy zprostředkované lipoproteinovou lipázou (LPL), a to zejména velkých chylomikronů nesoucích vysoké množství triglyceridů. Chylomikrony jsou vylučovány střevem po konzumaci jídla s obsahem tuku a z oběhu vymizí po 4–6 hodinách, proto je nelze detekovat nalačno. Vzácné bialelické mutace se ztrátou funkce v LPL nebo ve 4 dalších genech kódujících proteiny, které aktivují nebo interagují s LPL, jsou považovány za příčinu syndromu familiární chylomikronemie [38]. Příčiny monogenní chylomikronemie shrnuje tab. 2.
Syndrom monogenní chylomikronemie
Dosud bylo popsáno 5 genů, které se podílejí na katabolizmu triglyceridů v chylomikronech a jejichž bialelické mutace spojené se ztrátou funkce jsou příčinnou monogenní chylomikronemie – tj. LPL (kódující lipoproteinovou lipázu; LPL), APOC2 (kódující apolipoprotein C-II), APOA5 (kódující apolipoprotein AV), LMF1 (kódující faktor maturace lipázy 1,LMF1 ) a GPIHBP1 (kódující protein 1 ukotvený na glykosylfosfatidylinositolu, vázající HDL, GPIHBP1) [38–41]. Všechny tyto proteiny jsou nutné pro lipolýzu chylomikronů a VLDL zprostředkovanou LPL. Hladiny VLDL však mohou být normální, nebo nízké, protože sekrece VLDL je řízena převážně triglyceridy, které se do jater dostaly ve zbytcích chylomikronů. Sekrece VLDL může být zvýšena, pokud je přítomen také metabolický syndrom (tj. centrální obezita, inzulinová rezistence a diabetes). Více než 80 % jedinců s monogenní chylomikronemií má bialelické mutace LPL, kterých bylo identifikováno více než 100 [38,41].
Apolipoprotein CII je nezbytným koaktivátorem LPL. Ačkoliv bialelické mutace spojené se ztrátou funkce APOC2 způsobují fenotyp, který je v podstatě totožný s homozygotním deficitem LPL, molekulární vyšetření naznačují, že pouze 2–5 % jedinců s monogenní chylomikronemií má bialelické mutace APOC2 [41]. Podobně vzácná je úplná absence apolipoproteinu AV, o které se předpokládá, že usnadňuje interakci chylomikronů a VLDL s LPL na povrchu kapilárního endotelu. Bialelické mutace spojené se ztrátou funkce v APOA5 se vyskytují u 2–5 % jedinců s monogenní chylomikronemií [41], jejichž fenotyp je podobný deficitu LPL, avšak závažnost často závisí na sekundárních faktorech, jako je inzulinová rezistence nebo diabetes.
LMF1 je protein nezbytný pro správné sestavení a intracelulární přenos nascentní LPL a byl identifikován jako příčina kombinovaného deficitu lipáz u myší [42]. Deficit LMF1 vede k výrazně snížené sekreci LPL, což se projevuje těžkou hypertriglyceridemií podobně jako při deficitu LPL [42]. Bialelické mutace v LMF1 představují 1–2 % všech monogenních závažných hypertriglyceridemií [41].
A v poslední řadě GPIHBP1 přenáší LPL nově secernovanou přes kapilární endotel a stabilizuje enzym na povrchu endotelu, na němž interaguje s chylomikrony a VLDL [43]. Bialelické mutace v GPIHBP1, včetně rozsáhlých genových delecí, [41,44] způsobující úplný deficit GPIHBP1, jsou druhou nejčastější příčinou monogenní chylomikronemie, což odpovídá 5–10 % ze všech případů [41].
Monogenní chylomikronemie má podobnou závažnost v širokém spektru lipidových a metabolických fenotypů spojených s bialelickými mutacemi LPL, v porovnání s pacienty s mutacemi ve 4 menších genech [41]. Nadváha nebo inzulinová rezistence fenotyp dále zhoršují [38,41].
Další navrhované příčiny závažné hypetriglyceridemie
Úplná ztráta aktivity GPD1 (glycerol-3-fosfát dehydrogenázy 1) byla popsána u tranzitorní infantilní hypertriglyceridemie a pravděpodobně je důsledkem spíše zvýšené jaterní sekrece VLDL triglyceridů než chylomikronů [45]. Mezi další geny, jejichž mutace významně přispívají k těžké hypertriglyceridemii, patří CREB3L3, kódující transkripční faktor proteinu H, který se váže na cAMP responzivní element [46], a GCKR, kódující regulační protein glukokinázy [47]. Vzácné heterozygotní varianty se ztrátou funkce v těchto genech přispívají k polygenní predispozici, jak je popsáno v následujícím odstavci (Polygenní nebo multifaktoriální chylomikronemie). A konečně závažná hypertriglyceridemie je někdy sekundárním rysem vzácných monogenních forem inzulinové rezistence nebo diabetu, včetně familiárních generalizovaných nebo parciálních lipodystrofií.
Polygenní nebo multifaktoriální chylomikronemie
Mnoho klinických lékařů věří, že pacienti s těžkou hypertriglyceridemií musí mít monogenní poruchu. Nicméně příčinou závažné hypertriglyceridemie je nejčastěji interakce polygenní predispozice a sekundárních negenetických faktorů [6]. Například ve studii [39] zahrnující 563 pacientů s hladinou triglyceridů nad 10 mmol/l mělo pouze 6 (1,1 %) pacientů bialelické mutace v genech způsobujících monogenní chylomikronemie a 87 (15 %) pacientů bylo heterozygotními přenašeči mutace se ztrátou funkce v jednom z těchto genů oproti pouze 20 (4,0 %) pacientům z celkem 503 pacientů s normolipidemií. Ještě větší počet pacientů s těžkou hypertriglyceridemií má nadměrný počet běžných DNA polymorfizmů, z nichž každý zvyšuje koncentraci triglyceridů pouze o zlomek mmol/l. Někteří jedinci náhodou zdědí převahu polymorfizmů zvyšujících hladinu triglyceridů, které kumulativně zvyšují riziko vzniku těžké hyperglyceridemie. Například u pacientů s těžkou hypertriglyceridemií diskutovaných výše došlo u 180 (32 %) z 563 pacientů k extrémní akumulaci 32 běžných variant zvyšujících hladinu triglyceridů oproti 48 (9,5 %) ze 473 pacientů v kontrolní skupině [39]. Tato trojnásobně vyšší náchylnost na hypetriglyceridemii je typická pro polygenní dědičnost; riziko onemocnění u geneticky předisponovaných lidí je zvýšené, ale ne absolutně, protože část zdravých kontrol také nese stejnou genotypovou zátěž. U geneticky predisponovaných jedinců, u kterých se hypertriglyceridemie vyskytla, jsou často přítomny sekundární faktory.
Klinický obraz
Klinické projevy chylomikronemie shrnuje panel 1 a obr. 2. Těžká hypertriglyceridemie způsobená mutacemi spojenými se ztrátou funkce v 1 z 5 genů, které se podílejí na lipolýze, se často poprvé objeví už v dětství, dokonce už v kojeneckém věku a často se projevují špatným prospíváním a gastrointestinálními příznaky, jako je bolest břicha a pankreatitida [38]. Lipemický vzorek krve indikuje přítomnost akutní pankreatitidy vyvolané hypertriglyceridemií [38]. U starších dospívajících a dospělých, u kterých se časná pankreatitida neobjevila, může být diagnóza stanovena během rutinního vyšetření krve z jiných důvodů. Akutní pankreatitida může postihnout každého pacienta s koncentrací triglyceridů vyšší než 10 mmol/l. Zatímco relativní riziko je vyšší u monogenní chylomikronemie, v absolutních číslech je pankreatitida navozená hypertriglyceridemií mnohem častěji zjištěna u multifaktoriální nebo polygenní chylomikronemie [48]. Bez ohledu na genetické pozadí, závažnost hypertriglyceridemie (a tedy sklon k rozvoji pankreatitidy) zvyšuje konzumace potravin s vysokým obsahem tuku, alkohol, léky s obsahem estrogenů, těhotenství, obezita a inzulinová rezistence, diabetes, hypotyreóza nebo léky, které zvyšují sekreci VLDL (např. steroidy) [38]. Protože u všech genů platí, že oba rodiče budou heterozygoty, je povinný screening sourozenců postiženého dítěte, přičemž čtvrtina sourozenců bude mít také bialelické nebo homozygotní mutace. Fenotyp heterozygotních rodičů nebo sourozenců se může pohybovat v rozmezí od normálního lipidového profilu až po těžkou hypertriglyceridemii [39].

Diagnóza a léčba
Diagnóza monogenní chylomikronemie by měla být zvážena v případech, v nichž jsou koncentrace triglyceridů v plazmě vyšší než 10 mmol/l a zejména, když triglyceridy tuto hladinu výrazně překračují (obr. 5). Jak již bylo uvedeno, většina pacientů s takovými koncentracemi triglyceridů má multifaktoriální nebo polygenní chylomikronemii; podíl pacientů s monogenní chylomikronemií může být pouze 1–2 % [39]. Absence sekundárních faktorů a diagnóza ve velmi raném věku naznačují monogenní chylomikronemii, zejména jestliže hypertriglyceridemie způsobí pankreatitidu [48]. Nízké plazmatické hladiny apolipoproteinu B (< 0,75 g/l) mohou pomoci odlišit pacienty s monogenní a multifaktoriální chylomikronemií [48]. Těžká hypertriglyceridemie u sourozence v anamnéze také svědčí pro silný genetický podklad této poruchy. Klíčové však je odlišit monogenní těžkou hypertriglyceridemii od jiných komplexnějších příčin, jako je kombinace heterozygotní plus polygenní predispozice [39,48]. Genetické testování 5 genů zapojených do lipolýzy zprostředkované LPL a polygenní skóre pro hypertriglyceridemii mohou být užitečné pro objasnění genetické příčiny [7].
Terapie se zaměřuje na konzumaci nízkotučné stravy ideálně s obsahem méně než 10 % kalorií z tuku (panel 2). Dodržování tohoto režimu je však pro většinu pacientů velmi náročné. Použití mastných kyselin se středním řetězcem může poskytnout kalorie a esenciální mastné kyseliny a zároveň zabránit zvýšení koncentrací triglyceridů v plazmě [38]. Fibráty, které zvyšují aktivitu LPL, nejsou obvykle u pacientů s monogenní chylomikronemií prospěšné, ale mohou být účinné u pacientů s polygenní chylomikronemií. Vysoké dávky (4 g) omega-3-mastných kyselin, u nichž bylo prokázáno, že snižují koncentrace VLDL a pravděpodobně sekreci chylomikronů, mohou být účinné také u jedinců s polygenní hypertriglyceridemií. Malé množství přidaného tuku v potravě vyvažuje potenciální účinek této terapie [38].

Během epizody akutní pankreatitidy je úplné hladovění během několika prvních dnů léčby velmi účinné [49]. Důležité jsou i hydratace, analgezie a kontrola sekundárních faktorů; u pacientů s diabetem může být užitečná nitrožilní inzulinová terapie. Ačkoliv se v této situaci někdy doporučuje výměna plazmy, neexistují důkazy o tom, že by došlo ke zlepšení krátkodobých nebo dlouhodobých výsledků v porovnání s konzervativní léčbou [49]. Bez další metabolické léčby se navíc koncentrace triglyceridů rychle znovu obnoví. S možnou výjimkou léčby těžké hypertriglyceridemie způsobené monogenní chylomikronemií během těhotenství [50] se proto použití plazmaferézy nedoporučuje [49].
Limitace dostupných způsobů léčby jsou zřejmé, pacienti s monogenní chylomikronemií mají obvykle koncentrace triglyceridů vyšší než 20 mmol/l, a to i při dodržování správné diety a léčebného postupu. Riziko pankreatitidy je vždy přítomné a zapotřebí jsou účinnější terapie. Léčba ve vývoji včetně biologických látek, které snižují apolipoprotein CIII nebo ANGPTL3 , přináší možnost podstatného snížení koncentrací triglyceridů v plazmě u jedinců bez aktivity LPL z monogenních příčin [22]. Riziko trombocytopenie při léčbě monogenní chylomikronemie původním anti-APOC3 antisense přípravkem volanesorsenem je částečně sníženo u anti-APOC3 přípravku nové generace [51]. V Evropě byl volanesorsen schválen v roce 2019. Genová terapie LPL (alipogene tiparvovec) byla v Evropě schválena v roce 2012, ale výrobce po roce 2017 neobnovil licenci [22].
Dysbetalipiproteinemie
Dysbetalipoproteinemie (dříve známá jako choroba širokého β nebo hyperlipoproteinemie typu 3) postihuje 1 až 2 jedince na 20 000 lidí [52]. Triglyceridy i cholesterol jsou proměnlivě zvýšeny v důsledku patologické akumulace lipoproteinů se střední hustotou, neboli zbytků VLDL. Ačkoli se biochemicky podobá smíšené dyslipidemii, dysbetalipoproteinemii lze odlišit měřením koncentrací apolipoproteinu B [53]. Mezi typické klinické nálezy patří palmární a tuberoeruptivní xantomy na loktech a kolenou. Pacienti mají sklon k rozvoji předčasných koronárních onemocnění, a zejména k ischemické chorobě dolních končetin (ICHDK). Většina postižených jedinců jsou homozygoti pro izoformu APOE ε2, která kóduje protein s defektní vazbou na LDL-receptor, což vede k akumulaci zbytků chylomikronů s apolipoproteinem B48 v oběhu. Asi 10 % pacientů má v APOE dominantní variantu vzácné mutace se změnou smyslu (missense) s velkým účinkem. Jelikož se tyto genotypy vyskytují i u jedinců s normálním lipidogramem, jsou nutné další přispívající faktory včetně inzulinové rezistence nebo diabetu spolu se sekundárními negenetickými faktory (např. exogenní hormony, špatnou stravou, hypotyreózou, onemocněním ledvin, cukrovkou, paraproteinemií, nebo systémovým lupus erythematosus). Při léčbě je důležitá kompenzace sekundárních faktorů a léčba buď statiny, nebo fibráty, nebo obojí.
Monogenní hypotrigliceridemie
Dosud nebyl objeven žádný gen, který by sám mohl snížit pouze triglyceridy. Tato biochemická odchylka je typicky pouze složkou multisystémových poruch charakterizovaných nízkými až chybějícími lipoproteiny obsahujícími apolipoprotein B, jak jsou popsány výše, jako je abetalipoproteinemie, FHBL a deficit ANGPTL3. Deficit APOC3 se projevuje sníženými triglyceridy, zvýšeným HDL-cholesterolem a sníženým rizikem aterosklerotických kardiovaskulárních onemocnění [54]. Snížené hladiny triglyceridů mají u těchto stavů samy o sobě minimální až žádné klinické důsledky; léčba by se měla řídit obecnými doporučeními pro tyto poruchy.
Poruchy související s HDL
Plazmatické koncentrace HDL cholesterolu se rutinně v lipidovém panelu měří ze 2 hlavních důvodů: za prvé k odhadu koncentrací LDL-cholesterolu při absenci přímého měření [55] a za druhé k odhadu rizika kardiovaskulárních onemocnění podle důkazů z epidemiologických studií, že nízké koncentrace HDL-cholesterolu jsou spojeny se zvýšeným rizikem aterosklerotických kardiovaskulárních onemocnění [56]. HDL frakce zahrnují historickou třídu elektroforetické pohyblivosti lipoproteinů alfa.
Ačkoli přesná fyziologická role HDL není známa, konvenční přístup se zaměřuje na její přínos při zpětném transportu cholesterolu z makrofágů do jater (obr. 1) [57]. U lidí jsou však k dispozici minimální údaje, že by byl HDL mechanicky spojen s aterosklerózou a kardiovaskulárními příhodami; terapie cílené na HDL ve studiích neuspěly [57,58]. Negativní nález podpořily další práce vycházející z prospektivních kohort z běžné populace [59] a zhodnocení genetické zátěže pacientů s a bez infarktu myokardu [60]. Poznatky z epidemiologických studií pak naznačují složitější souvislost mezi HDL-cholesterolem a rizikem kardiovaskulárních příhod, chronickým onemocnění ledvin, infekcí a předčasným úmrtím, která je spíše ve tvaru písmene J nebo U než inverzní [61–64], s nejnižším rozmezím mezi 1,3–2,4 mmol/l podle pohlaví, etnika a komorbidit. Na základě těchto nových dat již neplatí, že HDL-cholesterol je ochranným faktorem pro celou populaci.
Poruchy spojené s nízkým HDL-cholesterolem (hypoalfalipoproteinemie)
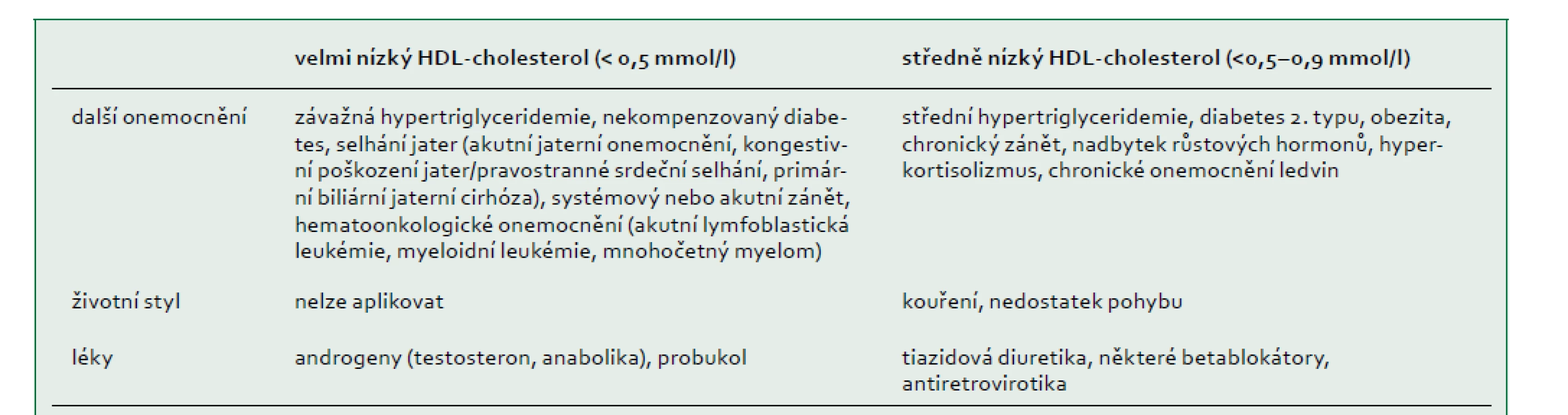
HDL-cholesterol má obvykle normální distribuci u žen a mužů s koncentracemi v extrémech buď v důsledku běžných sekundárních příčin (tab. 3), polygenních faktorů [65], nebo vzácných monogenních poruch. V americké studii s 258 252 jedinci, kterým bylo doporučeno vyšetření lipidů, mělo 504 (0,2 %) jedinců hladinu HDL-cholesterolu nižší než 0,52 mmol/l, což bylo způsobeno sekundární příčinou nalezenou u 206 (40 %) účastníků. Z 201 pacientů s identifikovaným genetickým podkladem bylo 14 (7 %) homozygotů, složených nebo dvojitých heterozygotů (tj. monogenních), zatímco 59 (29 %) bylo heterozygotů pro mutace genů APOA1, ABCA1, LCAT nebo LPL. U některých ze zbývajících 128 pacientů (64 %) byla možná polygenní predispozice [66].
Patofyziologie
Deficit apolipoproteinu AI a Tangierova choroba
I přes podobně nízké koncentrace HDL-cholesterolu a apolipoproteinu AI je klinický obraz deficitu apolipoproteinu AI a Tangierovy choroby odlišný v důsledku homozygotních mutací ABCA1, což naznačuje jednotlivé a orgánově specifické účinky ABCA1 na tvorbu HDL a homeostázy buněčného cholesterolu, jak bylo popsáno i ve studiích na zvířecích modelech [67–69]. Přestože apolipoprotein AI a ABCA1 v játrech a střevech mají klíčovou roli při produkci HDL, eflux cholesterolu zprostředkovaný ABCA1 brání vzniku pěnových buněk nezávisle na koncentraci HDL-cholesterolu v plazmě [70].
Deficit lecitin-cholesterol acyltransferázy a nemoc rybího oka
Familiární deficit lecitin-cholesterol acyltransferázy (LCAT) se projevuje absencí aktivity LCAT a absencí esterů cholesterolu v plazmě; neesterifikovaný cholesterol se v plazmě hromadí jako lipoprotein X (LpX), abnormální částice bohatá na cholesterol, která je vylučována hlavně retikuloendoteliálním systémem jater a sleziny [71]. U nemoci rybího oka LCAT ztrácí schopnost esterifikovat cholesterol na HDL, ale zachovává si svou aktivitu u LDL, což způsobuje nižší než normální hladinu esterů cholesterolu v plazmě [72].
Patogeneze onemocnění ledvin spojená s familiárním deficitem LCAT není zcela objasněna, ale v nichž poškozují endotel a cévy [74]. Jak familiární deficit LCAT, tak nemoc rybího oka se projevují nízkou koncentrací HDL-cholesterolu v plazmě a vadným zpětným transportem cholesterolu, a proto lze očekávat zvýšení kardiovaskulárního rizika. Při familiárním deficitu LCAT je však ateroskleróza potlačena. Tento pokles by mohl souviset se zachováním eliminace cholesterolu z makrofágů a nižšími koncentracemi LDL-cholesterolu u familiární deficience LCAT, ale nikoli u nemoci rybího oka [75].
Klinický obraz a diagnóza
Obr. 6 uvádí algoritmus pro diagnostiku a léčbu poruch spojených s velmi nízkými hladinami HDL-cholesterolu (tj. < 0,5 mmol/l) při absenci závažné hypertriglyceridemie. Před zvážením genetického podkladu je třeba nejprve vyloučit sekundární příčiny (tab. 3). Obecně se klinicky projevují pouze mutace se ztrátou funkce v homozygotní formě, nebo u složených heterozygotů v genech omezujících rychlost biogeneze HDL, i když existují i výjimky [76]. Specifické klinické příznaky mohou naznačit postup pro určení molekulární diagnózy (obr. 2).
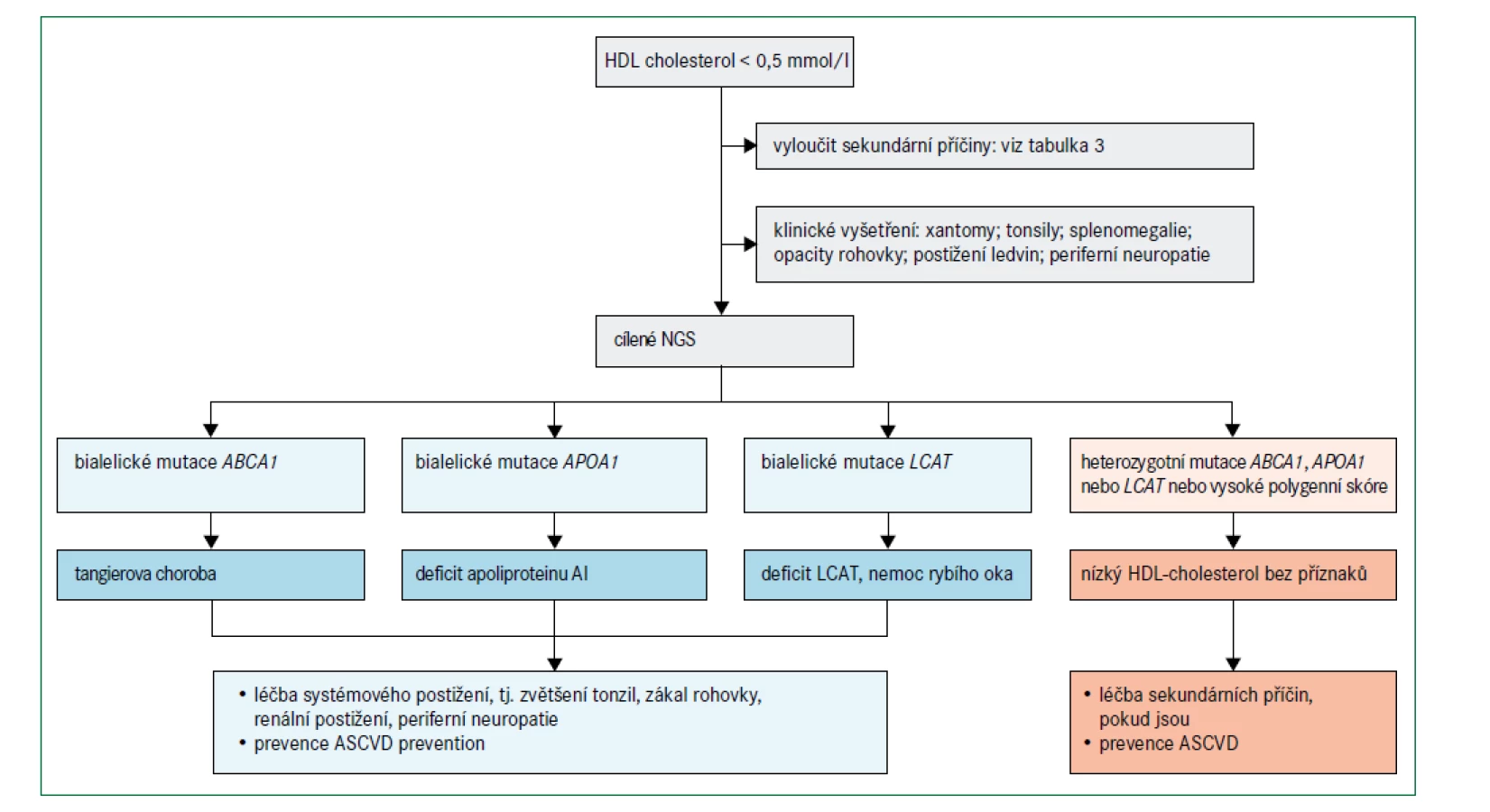
Mutace APOA1
Bylo popsáno více než 60 různých mutací se změnou smyslu (missense) v APOA1, přičemž data ze studie Copenhagen City Heart Study [77] popisují prevalenci heterozygotů přibližně 2,7 případů na 1 000 lidí. I přes nízké koncentrace HDL-cholesterolu jsou heterozygoti obvykle asymptomatičtí, ačkoli některé specifické ultra vzácné missense mutace jsou po transthyretinových variantách druhou nejčastější příčinou familiární amyloidózy [76]. Uvádí se, že umístění strukturální změny apolipoproteinu AI určuje místo ukládání amyloidu; ty mutace, které postihují aminoterminální doménu, jsou spojeny hlavně s jaterní a renální amyloidózou, zatímco mutace ovlivňující aminokyseliny 173–178 jsou většinou odpovědné za srdeční, laryngeální a kožní amyloidózu [76,78]. Pouze některé amyloidogenní varianty apolipoproteinu AI jsou spojeny s nízkou koncentrací HDL-cholesterolu; mnoho z nich bylo původně identifikováno imunohistochemickou analýzou amyloidu v postižených orgánech, ačkoli definitivní diagnóza nyní vyžaduje sekvenování genu APOA1 [7].
Deficit apolipoproteinu AI ve formě homozygotní, nebo jako složený heterozygot byl popsán u méně než 20 pacientů po celém světě a je charakterizován téměř nulovými hodnotami HDL-cholesterolu (< 0,3 mmol/l) a apolipoproteinu AI (< 0,1 g/l) a u většiny jedinců předčasnou ischemickou chorobou srdeční [79–81]. Pacienti se dvěma nulovými alelami mají xantomy, buď jen na očních víčkách, nebo až po celém těle (obr. 2) [79–81]. Pacienti s homozygotní nebo hemizygotní formou s mutacemi se změnou smyslu mají v plazmě reziduální koncentrace strukturně abnormálního apolipoproteinu AI a může u nich dojít k zákalu rohovky podobně jako u familiárního deficitu LCAT a nemoci rybího oka [79–81]. Tento příznak se však u pacientů s úplným deficitem apolipoproteinu AI neobjevuje vždy, někdy je detekovatelný pouze vyšetřením štěrbinovou lampou [79–81]. Definitivní diagnóza se stanovuje cíleným sekvenováním genu APOA1.
Tangierova choroba způsobená mutacemi ABCA1
Popsáno již bylo více než 170 mutací ABCA1 s odhadovanou prevalencí heterozygotů v populaci přibližně 3 případy na 1 000 lidí [59]. Diagnóza Tangierovy choroby spočívá v detekci bialelických mutací v ABCA1 [7,81–83], které způsobují velmi nízké plazmatické koncentrace HDL-cholesterolu a apolipoproteinu AI; v literatuře je popsáno více než 110 případů [81–83]. Klinický obraz je variabilní a souvisí s akumulací cholesterolu v makrofázích v různých orgánech s častými klinickými příznaky, jako jsou velké nažloutlé mandle (obr. 2), periferní neuropatie, splenomegalie, a hepatomegalie [81–83]. Mezi další laboratorní nálezy patří nízký počet krevních destiček, anémie, střední hypertriglyceridemie a nízké koncentrace LDL-cholesterolu [81–83]. Dosud není jasné, zda Tangierova choroba zvyšuje riziko aterosklerotických kardiovaskulárních onemocnění. Navzdory některým zprávám o předčasném infarktu myokardu u jedinců ve věku mezi 40 a 50 lety [81–83], jiní pacienti s Tangierovou chorobou, kteří zemřeli ve věku 60–70 let, neměli při pitvě žádné známky aterosklerózy [84]. Široké věkové rozpětí a zkreslení při analýzách komplikují určení rizika aterosklerotického kardiovaskulárního onemocnění; nízké koncentrace HDL-cholesterolu nejsou zcela vysvětlující. Zda má na klinický obraz a průběh onemocnění vliv určitá mutace, jiné faktory nebo kombinace obojího dosud není známo. Definitivní diagnóza je stanovena prokázáním bialelických mutací ABCA1 v sekvenci DNA.
Mutace LCAT u familiárního deficitu LCAT a nemoci rybího oka
Popsáno bylo více než 80 mutací genu LCAT [85], ale jejich prevalence v populaci není známa. Diagnóza familiárního deficitu LCAT a nemoci rybího oka je založena na biochemických parametrech a je omezena na nositele dvou mutantních alel LCAT. Obě poruchy se projevují velmi nízkými plazmatickými koncentracemi HDL-cholesterolu současně s nízkými koncentracemi LDL-cholesterolu a apolipoproteinu B, a to zejména u familiárního deficitu LCAT [81,85]. Opacita rohovky je častým projevem (obr. 2) a typicky se poprvé objeví během dospívání. Pacienti s familiárním deficitem LCAT mají také často mírnou chronickou normochromní anémii spojenou se zvýšeným počtem retikulocytů. Hlavní příčinou morbidity a mortality u pacientů s familiárním deficitem LCAT je onemocnění ledvin s proteinurií a progresivní renální insuficiencí [81,86,87], i když rychlost progrese je nepředvídatelná a variabilní. Definitivní diagnóza je stanovena prokázáním bialelických mutací v LCAT v sekvenci DNA.
Současná a budoucí léčba
V současnosti není k dispozici žádná specifická léčba deficitu apolipoproteinu AI a Tangierovy choroby; kyselina nikotinová (niacin) nebo fibráty u pacientů s těmito onemocněními hladiny HDL-cholesterolu nezvýší [88]. Pro léčbu komplikací periferní neuropatie je rovněž minimum možností. Základem ovlivnění rizika aterosklerotických kardiovaskulárních onemocnění je optimální kontrola nad dalšími rizikovými faktory včetně terapie snižující hladinu LDL-cholesterolu. Studie s podáváním infuze syntetického HDL po dobu 6 měsíců u 30 pacientů s deficitem apolipoproteinu AI nebo Tangierovou chorobou; neprokázala žádnou regresi aterosklerózy podle vyšetření pomocí 3-T MRI [89].
Podobně není k dispozici žádná specifická léčba syndromů deficitu LCAT. Publikováno bylo podávání inhibitorů enzymu konvertujícího angiotensin a antagonistů receptorů pro angiotenzin II pro snížení proteinurie a progrese onemocnění ledvin [90]. Závažné onemocnění ledvin vyžaduje hemodialýzu a nakonec i transplantaci ledvin, i když tato patologie se často rychle objeví znovu [91]. Po progresi opacity rohovky může být pro obnovení vidění nutná transplantace rohovky. Do budoucna by mohly mít potenciál nové přístupy, jako je enzymová substituční terapie lidským rekombinantním LCAT, nebo malé molekuly zvyšující aktivitu LCAT [92,93]. Po podání infuze rekombinantní LCAT byl u jednoho pacienta pozorován přínos na úrovni plazmatických lipidů, anémie a funkce ledvin [94].
Hyperalfalipoproteinemie
Hyperalphalipoproteinemie je způsobena mutacemi spojenými se ztrátou funkce v genu CETP, kódujícím transportní protein esterů cholesterolu (který zprostředkuje výměnu cholesterolu za triglyceridy a opačně mezi částicemi s apolipoproteinem B a HDL) [57,58], a mutacemi se ztrátou funkce v genu SRB1 (také známý jako SCARB1), kódujícím scavengerový receptor typu BI (SR-BI), což je jaterní receptor, který vychytává HDL pro produkci žluče (obr. 1). U obou příčin je hladina HDL-cholesterolu vyšší než 2,6 mmol/l [95–97]. Mutace obou genů působí kodominantně, přitom heterozygoti mají hladiny HDL-cholesterolu uprostřed mezi normou a homozygotem. Klinický fenotyp a riziko aterosklerotického kardiovaskulárního onemocnění nebylo u deficitu CETP přesně určeno a ještě méně informací máme o deficitu SR-BI, ačkoliv u některých pacientů byla popsána adrenální nedostatečnost, dysfunkce trombocytů [97,98] a také zvýšené riziko aterosklerotického kardiovaskulárního onemocnění [98]. Klinické studie, které hodnotily potenciál inhibitorů CETP při prevenci kardiovaskulárních příhod byly neprůkazné [99]. Další vzácnou monogenní příčinou hyperalfalipoproteinemie je deficit jaterní lipázy v důsledku bialelických mutací spojených se ztrátou funkce v genu LIPC, což vede ke kombinované dyslipidemii, a to hypercholesterolemii, hypertriglyceridemii a zvýšené koncentraci HDL s abnormálním složením [100]. Někteří z těchto pacientů mají zvýšené riziko aterosklerotického kardiovaskulárního onemocnění, a proto se u nich podávají statiny. V současnosti se nevyvíjí žádná nová léčiva na hyperalfalipoproteinemii, ale terapie se spíše zaměřuje na snížení rizika aterosklerotického kardiovaskulárního onemocnění pomocí současných způsobů léčby.
Způsob péče o pacienty
Péče o pacienty se vzácnými dyslipidemiemi by měla v ideálním případě probíhat ve specializovaném centru (např. tam, kde je k dispozici aferéza, pokud bude třeba). Péči by měl poskytovat zkušený lékař jako atestovaný lipidolog, endokrinolog, kardiolog, gastroenterolog nebo lékař primární péče. Odeslání pacienta k vyšetření a sledování jinou specializací je vhodné – např. oftalmolog pro pacienty s abetalipoproteinemií, FHBL, chorobou z retence chylomikronů nebo nemoci rybího oka; neurolog pro pacienty s abetalipoproteinemií, FHBL, chorobou z retence chylomikronů nebo Tangierovou chorobou; otolaryngolog pro pacienty s Tangierovou chorobou; a nefrolog pro pacienty s deficitem LCAT. Dětem by měla být poskytována péče od pediatra se zaměřením na dyslipidemie. Laboratorní vyšetření pacientů se vzácnými dyslipidemiemi je shrnuto v panelu 3. Webové stránky s informacemi pro poskytovatele péče a pacienty jsou uvedeny v panelu 4.


Závěr
Pokroky ve výzkumu genomu umožní v budoucnu využít výhody personalizované medicíny v léčbě vzácných dyslipidemií. Diagnóza založená na vyšetření DNA bude velmi rychlá a u vzácných dyslipidemií mnohem přesnější než předchozí diagnostická vyšetření (např. vyšetření aktivity enzymu nebo transportních proteinů z plazmy nebo ex vivo vyšetření aktivity receptoru, nebo efluxu cholesterolu). Až na několik výjimek (např. heterozygotní a homozygotní familiární hypercholesterolemie a monogenní chylomikronemie) neexistuje dosud žádný důkaz, že by diagnóza založená na vyšetření DNA byla určující nebo měla vliv na způsob terapie. Léčba pacientů s těmito vzácnými onemocněními se řídí zejména klinickými a biochemickými symptomy, přičemž možnosti léčby a přístupy vycházejí z observačních studií malých kohort pacientů a extrapolací rozsáhlejších studií. Nové způsoby léčby zacílené na klíčové molekulární dráhy byly nedávno schváleny nebo jsou pro vzácné dyslipidemie ve vývoji (tab. 4). V současnosti představuje u všech těchto poruch léčba založená na důkazech vzhledem k jejich nízké četnosti a interindividuální variabilitě příčin a exprese fenotypu výzvu pro kliniky. Tento konsensus pracovní skupiny EAS poskytuje snadno přístupné klinické doporučení pro lékaře, kteří usilují o zlepšení diagnostiky a zahájení příslušné léčby.
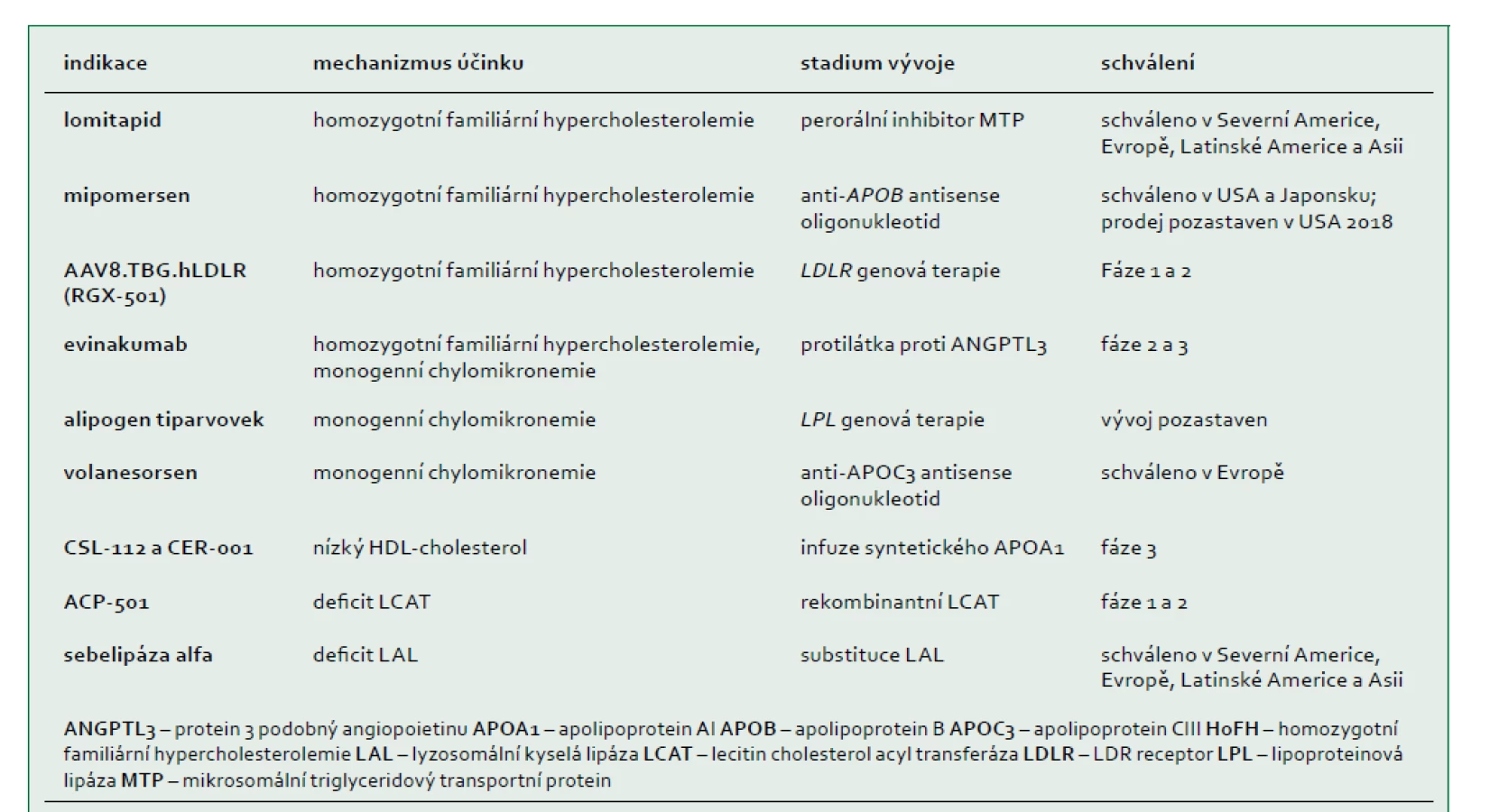
Pracovní skupina však identifikovala několik dosud nevyřešených problémů, jako jsou praktické obtíže v souvislosti s technologiemi a náklady a přístup k diagnostickým modalitám a nově vyvíjeným terapiím. Klíčovou slabinou je nedostatek informací o těchto poruchách, zejména s ohledem na prevalenci, patofyziologii a výsledný stav, jakož i absence účinných způsobů léčby jednotlivých poruch. Plátci zdravotní péče také požadují prospektivní data o klinické užitečnosti molekulární diagnostiky a kvalitní výsledky nových terapií, jejichž získání je logisticky náročné, protože celá globální populace jedinců se vzácnou dyslipidemií může čítat pouhých několik set nebo několik tisíc jedinců. Potenciálním geopolitickým problémem je zajištění přístupu k diagnostice a léčbě těchto převážně autosomálně recesivních poruch v regionech s vysokou prevalencí příbuznosti. Rozvoj registrů založených na integraci genomických technologií má potenciál přinést skutečný praktický užitek všem zúčastněným stranám. Mezi tyto výhody patří zlepšení informovanosti, poskytování péče a přístupu k účinné terapii těchto poruch. Farmaceutický průmysl by mohl v budoucnu hrát klíčovou roli při spolupráci s vládami a nevládními organizacemi. Komplementární a koordinovaná politická, ekonomická a socioekonomická opatření by v kombinaci s technologickým pokrokem mohly zmírnit „poddiagnostikování“ a nedostatečnou léčbu a nakonec změnit zdravotní péči o pacienty se vzácnými poruchami lipoproteinů.

Přispěvatelé
Této pracovní skupině Evropské spol. pro aterosklerózu společně předsedali ALC a HNG. Jednotlivé části byly koncipovány třemi skupinami, které se zaměřily na LDL-cholesterol (ALC, MAv), JB, MC, FK, KGP, FJR, KKR, JKS, LT); triglycerid (HNG, RAH, MAr, DG,ESS) a HDL (CJB, LC avE, RF-S, GKH, DL aTR). Návrh byl posouzen spolupředsedajícími, JB, MJC a RAH. Všichni autoři před podáním přezkoumali a schválili finální rukopis.
Prohlášení o zájmech
RAH získal granty a honoráře za poradenství od spol. Acasti, Akcea a Ionis, granty od spol. Regeneron a Boston Heart Diagnostics a honoráře za poradenství od spol. Aegerion, Amgen, Gemphire a Sanofi. JB získal granty od spol. AstraZeneca a Pfizer, granty a honoráře za poradenství od spol. Amgen, NovoNordisk, Regeneron a Sanofi a honoráře za poradenství od spol. Akcea, Eli Lilly a Merck. HNG získala granty a honoráře za poradenství od spol. Merck, granty od spol. Sanofi-Regeneron, Amgen, Medimmune a AstraZeneca a honoráře za poradenství od spol. Janssen, Sanofi, Regeneron, Kowa, Pfizer a Resverlogix. MAr získala granty od spol. Aegerion a Regeneron, granty a honoráře za poradenství od spol. Akcea, Ionis, Amgen, Amyrt a Sanofi a honoráře za poradenství od spol. Alfasigma, Mylan a Pfizer. MAv získala granty a honoráře za poradenství od spol. Aegerion, Akcea, Ionis, Alfasigma, Amgen, Amryt, Pfizer, Regeneron a Sanofi. CJB získala honoráře za poradenství od spol. Amgen. LC získala granty a honoráře za poradenství od spol. MedImmune a granty od spol. Cerenis therapeutics, Daiichi Sankyo a Alexion Pharma. MJC získal granty od spol. Amgen, Kowa Europe a Pfizer a honoráře za přednášky a zprostředkování řečníků od spol. Akcea, Alexion, Amarin, Amgen, Daiichi-Sankyo, Kowa Europe, Merck, MSD, Pfizer, Sanofi, Regeneron a Unilever. MC získala granty od spol. RegenXBio, Regeneron Pharmaceuticals a Akcea Therapeutics. Společnost DG získala granty a honoráře za přednášky a zprostředkování řečníků nebo poradenství od spol. Aegerion, Akcea, Amgen, HDL Therapeutics, Ionis Pharmaceuticals, Novartis, Regeneron a Sanofi, granty od spol. Acasti, AstraZeneca, Boehringer Ingelheim, Canadian Cardiovascular Research Network, Cerenis, Dalcor Pharma, Esperion, Gemphire, GlaxoSmithKline, Institut de Cardiologie de Montreal, Ironwood, Kowa, Lilly, Pfizer, The Medicines Company a UniQure a honoráře za přednášky a zprostředkování řečníků nebo poradenství od spol. Nestlé. GKH získala granty od organizací Netherlands Organisation for Scientific Research, CardioVascular Research Initiative a Evropské unie, honoráře za poradenství a nefinanční podporu od spol. Amgen, Aegerion, AstraZeneca a Sanofi, honoráře za přednášky a zprostředkování řečníků nebo poradenství od spol. Pfizer, Regeneron, Kowa, Ionis Pharmaceuticals a Cerenis a nefinanční podporu od spol. Synageva. KGP získala granty a honoráře za poradenství od spol. Sanofi, granty od spol. Novartis a honoráře za poradenství od spol. Amgen, MSD, Akcea, Silence Therapeutics, Daiichi Sankyo a Regeneron. FJR získala honoráře za poradenství a nefinanční podporu od spol. Amgen, Sanofi, Regeneron a The Medicines Company. KKR získala granty a honoráře za poradenství, poradní výbory a přednášky od spol. Amgen, Sanofi, Regeneron, MSD a Pfizer, honoráře za poradenství, poradní výbory a přednášky od spol. AbbVie, AstraZeneca, The Medicines Company, Resverlogix, Akcea, Boehringer Ingelheim, Novo Nordisk, Takeda, Kowa, Algorithm, Cipla, Cerenis, Dr. Reddys, Lilly, Zuellig Pharma, Silence Theapeutics a Bayer. ESS získala honoráře za přednášky a zprostředkování řečníků od spol. Amgen, Sanofi, Akcea a Novartis. LT získala honoráře za přednášky a zprostředkování řečníků nebo poradenství od spol. MSD, Sanofi, Amgen, Abbott, Mylan, Bayer, Actelion, Novartis, Astra, Recordati, Pfizer, Servier a Novo Nordisk. Je také předsedkyní Evropské spol. pro aterosklerózu a členkou redakční rady časopisu European Heart Journal. ALC získala granty od společností Pfizer, Sanofi, Regeneron, Merck a Mediolanum, nefinanční podporu od spol. SigmaTau, Menarini, Kowa, Recordati a Eli Lilly a honoráře za přednášky a zprostředkování řečníků nebo poradenství od spol. AstraZeneca, Genzyme, Menarini, Kowa, El Lilly, Recordati, Pfizer, Sanofi, Mediolanum, Pfizer, Merck, Sanofi, Aegerion a Amgen.
Všichni ostatní autoři prohlásili, že nemají střet zájmů.
Poděkování
Členové pracovní skupiny EAS se setkali dvakrát na uzavřených jednáních odborníků v Amsterodamu (5.–6. 7. 2018) a Mnichově (16.–17. 10. 2018), aby kriticky zhodnotili a prodiskutovali důkazy týkající se prevalence, patofyziologie, prezentace a péče o vzácné poruchy metabolizmu lipoproteinů, které se vyznačují extrémními koncentracemi lipidů (LDL-cholesterolu, triglyceridů a HDL-cholesterolu). Logistickou podporu cestování poskytla Evropská společnost pro aterosklerózu. Žádné jiné zdroje financování nebyly použity.
Lancet Diabetes Endocrinol 2019
Published Online September 30, 2019 https://doi.org/10.1016/ S2213-8587(19)30264-5
Correspondence to:
Prof Robert A Hegele
Robarts Research Institute, Western University, London, ON N6A 5C1, Canada
garant prof. MUDr. Michal Vrablík, Ph.D., předseda ČSAT
www.athero.cz
Doručeno do redakce | Doručené do redakcie | Received 25. 3. 2021
Zdroje
1 Richter T, Nestler-Parr S, Babela R, et al. Rare disease terminology and definitions—a systematic global review: report of the ISPOR rare disease special interest group. Value Health 2015; 18: 906–14.
2 European Medicines Agency. Orphan designation: overview. https://www.ema.europa.eu/en/human-regulatory/overview/ orphan-designation-overview (accessed April 4, 2019).
3 Orphan Drug Act. Rare diseases act of 2002. https://www.govinfo. gov/content/pkg/PLAW-107publ280/html/PLAW-107publ280.htm (accessed April 4, 2019).
4 EURORDIS. Fact sheet. What is a rare disease? https://www. eurordis.org/sites/default/files/publications/Fact_Sheet_RD.pdf (accessed April 9, 2019).
5 Boycott KM, Hartley T, Biesecker LG, et al. A diagnosis for all rare genetic diseases: the horizon and the next frontiers. Cell 2019; 177: 32–37.
6 Dron JS, Hegele RA. Polygenic influences on dyslipidemias. Curr Opin Lipidol 2018; 29: 133–43.
7 Hegele RA, Ban MR, Cao H, McIntyre AD, Robinson JF, Wang J. Targeted next-generation sequencing in monogenic dyslipidemias. Curr Opin Lipidol 2015; 26: 103–13.
8 Hegele RA. Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet 2009; 10: 109–21.
9 Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 2013; 34: 3478–90a.
10 Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers 2017; 3: 17093.
11 Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the consensus panel on familial hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J 2014; 35: 2146–57.
12 Arca M, Zuliani G, Wilund K, et al. Autosomal recessive hypercholesterolaemia in Sardinia, Italy, and mutations in ARH: a clinical and molecular genetic analysis. Lancet 2002; 359: 841–47.
13 Iacocca MA, Chora JR, Carrié A, et al. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum Mutat 2018; 39: 1631–40.
14 Andersen LH, Miserez AR, Ahmad Z, Andersen RL. Familial defective apolipoprotein B-100: a review. J Clin Lipidol 2016; 10: 1297–302.
15 Dron JS, Hegele RA. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr Opin Lipidol 2017; 28: 161–69.
16 Wang J, Dron JS, Ban MR, et al. Polygenic versus monogenic causes of hypercholesterolemia ascertained clinically. Arterioscler Thromb Vasc Biol 2016; 36: 2439–45.
17 Berberich AJ, Hegele RA. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol 2019; 16: 9–20.
18 Reiner Ž, Guardamagna O, Nair D, et al. Lysosomal acid lipase deficiency—an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 2014; 235: 21–30.
19 Julius U. Current role of lipoprotein apheresis in the treatment of high-risk patients. J Cardiovasc Dev Dis 2018; 5: e27.
20 Raal FJ, Hovingh GK, Blom D, et al. Long-term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open-label TAUSSIG study. Lancet Diabetes Endocrinol 2017; 5: 280–90.
21 Blom DJ, Averna MR, Meagher EA, et al. Long-term efficacy and safety of the microsomal triglyceride transfer protein inhibitor lomitapide in patients with homozygous familial hypercholesterolemia. Circulation 2017; 136: 332–35.
22 Hegele RA, Tsimikas S. Lipid-lowering agents. Circ Res 2019; 124: 386–404.
23 Gaudet D, Gipe DA, Pordy R, et al. ANGPTL3 inhibition in homozygous familial hypercholesterolemia. N Engl J Med 2017; 377: 296–97.
24 Kidambi S, Patel SB. Sitosterolaemia: pathophysiology, clinical presentation and laboratory diagnosis. J Clin Pathol 2008; 61: 588–94.
25 Burton BK, Balwani M, Feillet F, et al. A phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med 2015; 373: 1010–20.
26 Hooper AJ, Burnett JR. Update on primary hypobetalipoproteinemia. Curr Atheroscler Rep 2014; 16: 423.
27 Hooper AJ, van Bockxmeer FM, Burnett JR. Monogenic hypocholesterolaemic lipid disorders and apolipoprotein B metabolism. Crit Rev Clin Lab Sci 2005; 42: 515–45.
28 Lee J, Hegele RA. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: a framework for diagnosis and management. J Inherit Metab Dis 2014; 37: 333–39.
29 Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol 2014; 25: 161–68.
30 Parhofer KG, Barrett PH, Bier DM, Schonfeld G. Lipoproteins containing the truncated apolipoprotein, Apo B-89, are cleared from human plasma more rapidly than Apo B-100-containing lipoproteins in vivo. J Clin Invest 1992; 89: 1931–37.
31 Welty FK, Lichtenstein AH, Barrett PH, Dolnikowski GG, Ordovas JM, Schaefer EJ. Decreased production and increased catabolism of apolipoprotein B-100 in apolipoprotein B-67/B-100 heterozygotes. Arterioscler Thromb Vasc Biol 1997; 17: 881–88.
32 Parhofer KG, Barrett PH, Aguilar-Salinas CA, Schonfeld G. Positive linear correlation between the length of truncated apolipoprotein B and its secretion rate: in vivo studies in human apoB-89, apoB-75, apoB-54.8, and apoB-31 heterozygotes. J Lipid Res 1996; 37: 844–52.
33 Levy E, Poinsot P, Spahis S. Chylomicron retention disease: genetics, biochemistry, and clinical spectrum. Curr Opin Lipidol 2019; 30: 134–39.
34 Minicocci I, Montali A, Robciuc M R, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab 2012; 97: e1266–75.
35 Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med 2010; 363: 2220–27.
36 Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006; 79: 514–23.
37 Di Costanzo A, Di Leo E, Noto D, et al. Clinical and biochemical characteristics of individuals with low cholesterol syndromes: a comparison between familial hypobetalipoproteinemia and familial combined hypolipidemia. J Clin Lipidol 2017; 11: 1234–42.
38 Brahm AJ, Hegele RA. Chylomicronaemia—current diagnosis and future therapies. Nat Rev Endocrinol 2015; 11: 352–62.
39 Dron JS, Wang J, Cao H, et al. Severe hypertriglyceridemia is primarily polygenic. J Clin Lipidol 2019; 13: 80–88.
40 Dron JS, Hegele RA. Genetics of triglycerides and the risk of atherosclerosis. Curr Atheroscler Rep 2017; 19: 31.
41 Hegele RA, Berberich AJ, Ban MR, et al. Clinical and biochemical features of different molecular etiologies of familial chylomicronemia. J Clin Lipidol 2018; 12: 920–27.
42 Péterfy M. Lipase maturation factor 1: a lipase chaperone involved in lipid metabolism. Biochim Biophys Acta 2012; 1821: 790–94.
43 Fong LG, Young SG, Beigneux AP, et al. GPIHBP1 and plasma triglyceride metabolism. Trends Endocrinol Metab 2016; 27: 455–69.
44 Iacocca MA, Dron JS, Hegele RA. Progress in finding pathogenic DNA copy number variations in dyslipidemia. Curr Opin Lipidol 2019; 30: 63–70.
45 Basel-Vanagaite L, Zevit N, Har Zahav A, et al. Transient infantile hypertriglyceridemia, fatty liver, and hepatic fibrosis caused by mutated GPD1, encoding glycerol-3-phosphate dehydrogenase 1. Am J Hum Genet 2012; 90: 49–60.
46 Lee JH, Giannikopoulos P, Duncan SA, et al. The transcription factor cyclic AMP-responsive element-binding protein H regulates triglyceride metabolism. Nat Med 2011; 17: 812–15.
47 Rees MG, Raimondo A, Wang J, et al. Inheritance of rare functional GCKR variants and their contribution to triglyceride levels in families. Hum Mol Genet 2014; 23: 5570–78.
48 Paquette M, Bernard S, Hegele RA, Baass A. Chylomicronemia— differences between familial chylomicronemia syndrome and multifactorial chylomicronemia. Atherosclerosis 2019; 283: 137–42.
49 Berberich AJ, Ziada A, Zou GY, Hegele RA. Conservative management in hypertriglyceridemia-associated pancreatitis. J Intern Med 2019; published online May 11. DOI:10.1111/joim.12925.
50 Goldberg AS, Hegele RA. Severe hypertriglyceridemia in pregnancy. J Clin Endocrinol Metab 2012; 97: 2589–96.
51 Alexander VJ, Xia S, Hurh E, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J 2019; published online April 24. DOI:10.1093/eurheartj/ehz209.
52 Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology 2019; 51: 165–76.
53 Sniderman AD, de Graaf J, Thanassoulis G, Tremblay AJ, Martin SS, Couture P. The spectrum of type III hyperlipoproteinemia. J Clin Lipidol 2018; 12: 1383–89.
54 Pollin T, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008; 322: 1702–05.
55 Langlois MR, Chapman MJ, Cobbaert C, et al. Quantifying atherogenic lipoproteins: current and future challenges in the era of personalized medicine and very low concentrations of LDL cholesterol. A consensus statement from EAS and EFLM. Clin Chem 2018; 64: 1006–33.
56 Catapano AL, Graham I, De Backer G, et al. 2016 ESC/EAS Guidelines for the management of dyslipidaemias. Eur Heart J 2016; 37: 2999–3058.
57 März W, Kleber ME, Scharnagl H, Speer T, et al. HDL cholesterol: reappraisal of its clinical relevance. Clin Res Cardiol 2017; 106: 663–75.
58 Annema W, von Eckardstein A. High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J 2013; 77: 2432–48.
59 Frikke-Schmidt R, Nordestgaard BG, Stene MC, et al. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 2008; 299: 2524–32.
60 Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012; 380: 572–-80.
61 Bowe B, Xie Y, Xian H, Balasubramanian S, Zayed MA, Al-Aly Z. High density lipoprotein cholesterol and the risk of all-cause mortality among US veterans. Clin J Am Soc Nephrol 2016; 11: 1784–93.
62 Ko DT, Alter DA, Guo H, et al. High-density lipoprotein cholesterol and cause-specific mortality in individuals without previous cardiovascular conditions: the CANHEART Study. J Am Coll Cardiol 2016; 68: 2073–83.
63 Madsen CM, Varbo A, Nordestgaard BG. Extreme high high-density lipoprotein cholesterol is paradoxically associated with high mortality in men and women: two prospective cohort studies. Eur Heart J 2017; 38: 2478–86.
64 Madsen CM, Varbo A, Tybjærg-Hansen A, Frikke-Schmidt R, Nordestgaard BG. U-shaped relationship of HDL and risk of infectious disease: two prospective population-based cohort studies. Eur Heart J 2018; 39: 1181–90.
65 Dron JS, Wang J, Low-Kam C, et al. Polygenic determinants in extremes of high-density lipoprotein cholesterol. J Lipid Res 2017; 58: 2162–70.
66 Geller AS, Polisecki EY, Diffenderfer MR, et al. Genetic and secondary causes of severe HDL deficiency and cardiovascular disease. J Lipid Res 2018; 59: 2421–35.
67 Timmins JM, Lee JY, Boudyguina E, et al. Targeted inactivation of hepatic ABCA1 causes profound hypoalphalipoproteinemia and kidney hypercatabolism of ApoA-I. J Clin Invest 2005; 115: 1333–42.
68 Brunham LR, Kruit JK, Iqbal J, et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J Clin Invest 2006; 116: 1052–62.
69 Zhu X, Lee JY, Timmins JM, et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J Biol Chem 2008; 283: 22930–41.
70 Rosenson RS, Brewer HB Jr, Davidson WS, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation 2012; 125: 1905–19.
71 Fellin R, Manzato E. Lipoprotein-X fifty years after its original discovery. Nutr Metab Cardiovasc Dis 2019; 29: 4–8.
72 Calabresi L, Simonelli S, Gomaraschi M, Franceschini G. Genetic lecithin: cholesterol acyltransferase deficiency and cardiovascular disease. Atherosclerosis 2012; 222: 299–306.
73 Ossoli A, Neufeld EB, Thacker SG, et al. Lipoprotein X causes renal disease in LCAT deficiency. PLoS One 2016; 11: e0150083.
74 Imbasciati E, Paties C, Scarpioni L, Mihatsch MJ. Renal lesions in familial lecithin-cholesterol acyltransferase deficiency. Ultrastructural heterogeneity of glomerular changes. Am J Nephrol 1986; 6: 66–70.
75 Oldoni F, Baldassarre D, Castelnuovo S, et al. Complete and partial LCAT deficiency are differentially associated with atherosclerosis. Circulation 2018; 138: 1000–07.
76 Lu C, Zuo K, Lu Y, et al. Apolipoprotein A-1-related amyloidosis: 2 case reports and review of the literature. Medicine (Baltimore) 2017; 96: e8148.
77 Haase CL, Frikke-Schmidt R, Nordestgaard BG, Tybjærg-Hansen A. Population-based resequencing of APOA1 in 10,330 individuals: spectrum of genetic variation, phenotype, and comparison with extreme phenotype approach. PLoS Genet 2012; 8: e1003063.
78 Das M, Wilson CJ, Mei X, Wales TE, Engen JR, Gursky O. Structural stability and local dynamics in disease-causing mutants of human apolipoprotein A-I: what makes the protein amyloidogenic? J Mol Biol 2016; 428: 449–62.
79 Santos RD, Asztalos BF, Martinez LR, Miname MH, Polisecki E, Schaefer EJ. Clinical presentation, laboratory values, and coronary heart disease risk in marked high-density lipoprotein-deficiency states. J Clin Lipidol 2008; 2: 237–47.
80 Tanaka S, Haketa A, Sakimoto T, Abe M. A case of apolipoprotein A-I deficiency due to carboxyl-terminal truncation. J Clin Lipidol 2018; 12: 511–14.
81 Schaefer EJ, Anthanont P, Diffenderfer MR, Polisecki E, Asztalos BF. Diagnosis and treatment of high density lipoprotein deficiency. Prog Cardiovasc Dis 2016; 59: 97–106.
82 Hooper AJ, McCormick SPA, Hegele RA, Burnett JR. Clinical utility gene card for: Tangier disease. Eur J Hum Genet 2017; published online May 24. DOI:10.1038/ejhg.2017.72.
83 Muratsu J, Koseki M, Masuda D, et al. Accelerated atherogenicity in Tangier Disease. J Atheroscler Thromb 2018; 25: 1076–85.
84 Serfaty-Lacrosniere C, Civeira F, Lanzberg A, et al. Homozygous Tangier disease and cardiovascular disease. Atherosclerosis 1994; 107: 85–98.
85 Saeedi R, Li M, Frohlich J. A review on lecithin: cholesterol acyltransferase deficiency. Clin Biochem 2015; 48: 472–75.
86 Jonas A. Lecithin cholesterol acyltransferase. Biochem Biophys Acta 2000; 1529: 245–46.
87 Holleboom AG, Kuivenhoven JA, van Olden CC, et al. Proteinuria in early childhood due to familial LCAT deficiency caused by loss of a disulfide bond in lecithin:cholesterol acyl transferase. Atherosclerosis 2011; 216: 161–65.
88 Alrasadi K, Awan Z, Alwaili K, et al. Comparison of treatment of severe high-density lipoprotein cholesterol deficiency in men with daily atorvastatin (20 mg) versus fenofibrate (200 mg) versus extended-release niacin (2 g). Am J Cardiol 2008; 102: 1341–47.
89 Kootte RS, Smits LP, van der Valk FM, et al. Effect of open-label infusion of an apoA-I-containing particle (CER-001) on RCT and artery wall thickness in patients with FHA. J Lipid Res 2015; 56: 703–12.
90 Aranda P, Valdivielso P, Pisciotta L, et al. Therapeutic management of a new case of LCAT deficiency with a multifactorial long-term approach based on high doses of angiotensin II receptor blockers (ARBs). Clin Nephrol 2008; 69: 213–18.
91 Strom EH, Sund S, Reier-Nilsen M, Dorje C, Leren TP. Lecithin: cholesterol acyltransferase (LCAT) deficiency: renal lesions with early graft recurrence. Ultrastruct Pathol 2011; 35: 139–45.
92 Shamburek RD, Bakker-Arkema R, Shamburek AM, et al. Safety and tolerability of ACP-501, a recombinant human lecithin: cholesterol acyltransferase, in a phase 1 single-dose escalation study. Circ Res 2016; 118: 73–82.
93 Freeman LA, Demosky SJ Jr, Konaklieva M, et al. Lecithin: cholesterol acyltransferase activation by sulfhydryl-reactive small molecules: role of cysteine-31. J Pharmacol Exp Ther 2017; 362: 306–18.
94 Shamburek RD, Bakker-Arkema R, Auerbach BJ, et al. Familial lecithin: cholesterol acyltransferase deficiency: first-in-human treatment with enzyme replacement. J Clin Lipidol 2016; 10: 356–67.
95 Inazu A, Brown ML, Hesler CB, et al. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med 1990; 323: 1234–38.
96 Vergeer M, Korporaal SJ, Franssen R, et al. Genetic variant of the scavenger receptor BI in humans. N Engl J Med 2011; 364: 136–45.
97 Brunham LR, Tietjen I, Bochem AE, et al. Novel mutations in scavenger receptor BI associated with high HDL cholesterol in humans. Clin Genet 2011; 79: 575–81.
98 Zanoni P, Khetarpal SA, Larach DB, et al. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016; 351: 1166–71.
99 Tall AR, Rader DJ. Trials and tribulations of CETP Inhibitors.
Circ Res 2018; 122: 106–12.
100 Hegele RA, Little JA, Vezina C, et al. Hepatic lipase deficiency. Clinical, biochemical, and molecular genetic characteristics.
Arterioscler Thromb 1995; 13: 720–08.
101 Davignon J, Dufour R. Primary hyperlipidemias. Oxford: Clinical Publishing, 2007.
102 Genest J, Hegele RA, Bergeron J, et al. Canadian Cardiovascular Society position statement on familial hypercholesterolemia. Can J Cardiol 2014; 30: 1471–81.
103 Hamel C. Retinitis pigmentosa. Orphanet J Rare Dis 2006; 1: 40.
104 Sassolas A, Filippo M Di, Aggerbeck LP, Peretti N, Samson-Bouma ME. Anderson’s disease/chylomicron retention disease and mutations in the SAR1B gene, mutations in human genetic disease. London: IntechOpen, 2012.
105 Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ 2007; 176: 1113–20.
106 Ravesloot MJ, Bril H, Braamskamp MJ, Wiegman A, Wong Chung RP. The curious case of the orange coloured tonsils. Int J Ped Otorhinolaryngology 2014; 78: 2305–07.
Štítky
Angiologie Diabetologie Interní lékařství Kardiologie Praktické lékařství pro dospěléČlánek vyšel v časopise
Athero Review

2021 Číslo Supplementum 1
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- Antidepresiva skupiny SSRI v rukách praktického lékaře
Nejčtenější v tomto čísle
