
Doporučení pro diagnostiku a léčbu chronické lymfocytární leukemie (CLL) – 2018
Recommendations for the diagnosis and treatment of chronic lymphocytic leukaemia (CLL) – 2018
Chronic lymphocytic leukaemia has a remarkably heterogeneous clinical course. The diagnostic procedures and therapeutic interventions need to be individually tailored according to patient age, comorbidities and therapeutic aims. There have been crucial developments in prognostication and therapy in recent years (e.g., significance of TP53 mutations, introduction of novel monoclonal antibodies, B-cell receptor signalling and Bcl-2 inhibitors). Therefore, the Czech CLL Study Group, the working group of the Czech Haematological Society, has developed these updated guidelines to facilitate the decision-making process for diagnosis and treatment in clinical practice. The guidelines are based on a comprehensive analysis of current literature and the principles of evidence-based medicine.
Key words:
chronic lymphocytic leukemia – diagnostics – treatment
Czech Group for Chronic Lymphocytic Leukaemia, section of the Czech Society of Haematology of the Czech medical Society of Jan Evangelista Purkyně
Autoři:
M. Doubek 1; M. Špaček 2; Š. Pospíšilová 1; M. Jarošová 1; T. Papajík 3; R. Urbanová 3; M. Šimkovič 4; D. Lysák 5; M. Brejcha 6; L. Smolej 4
Působiště autorů:
Interní hematologická a onkologická klinika, Fakultní nemocnice a Lékařská fakulta Masarykovy Univerzity Brno
1; I. interní klinika – klinika hematologie, 1. lékařská fakulta Univerzity Karlovy a Všeobecná fakultní nemocnice Praha
2; Hemato-onkologická klinika, Fakultní nemocnice a Lékařská fakulta Univerzity Palackého Olomouc
3; IV. interní hematologická klinika, Fakultní nemocnice Hradec Králové a Lékařská fakulta Univerzity Karlovy v Hradci Králové
4; Hematologicko-onkologické oddělení, Fakultní nemocnice Plzeň
5; Hematologické oddělení, Onkologické centrum Nový Jičín
6
Vyšlo v časopise:
Transfuze Hematol. dnes,24, 2018, No. 3, p. 208-220.
Kategorie:
Doporučené postupy
Souhrn
Chronická lymfocytární leukemie je onemocnění s mimořádně různorodým klinickým průběhem. Diagnostiku a léčbu je nutno individualizovat s přihlédnutím k věku, celkovému stavu, přidruženým chorobám a cílům léčby. V posledních letech přibyly zásadní poznatky týkající se posouzení prognózy i léčby (význam mutací genu TP53, nové monoklonální protilátky, inhibitory drah B-buněčného receptoru, inhibitory Bcl-2 a další). Česká skupina pro CLL, sekce České hematologické společnosti ČLS JEP, proto vytvořila tato aktualizovaná doporučení k usnadnění rozhodování o diagnostických a léčebných postupech v klinické praxi. Doporučení se zakládají na důkladné analýze současné literatury a vycházejí z principů medicíny založené na důkazech.
Klíčová slova:
chronická lymfocytární leukemie – diagnostika – léčba
Česká skupina pro chronickou lymfocytární leukemii (ČSCLL), sekce České hematologické společnosti České lékařské společnosti Jana Evangelisty Purkyně (ČHS ČLS JEP)
1. Stanovení diagnózy chronické lymfocytárníleukemie (CLL)
Diagnóza CLL je stanovena podle doporučení International Workshop on CLL (IWCLL) na základě vyšetření krevního obrazu s mikroskopickým rozpočtem a průtokové cytometrie periferní krve [1]. Diagnostika vyžaduje přítomnost minimálně 5 x 109/l B lymfocytů v periferní krvi s průkazem charakteristického fenotypu a klonality průtokovou cytometrií. V krevním nátěru jsou nacházeny převážně malé, zralé lymfocyty s úzkým lemem cytoplazmy a kondenzovaným jádrem bez jadérka; může se vyskytovat příměs větších, atypických lymfocytů s naštípnutým jádrem nebo prolymfocytů, které mohou tvořit až 55 % všech leukemických buněk (tab. 1). Nález prolymfocytů nad 55 % vede společně s nálezem v průtokové cytometrii k diagnóze B-prolymfocytární leukemie (B-PLL) [1, 2]. Vzhledem k tomu, že maligní klon je možno jednoznačně identifikovat v periferní krvi, není pro stanovení diagnózy CLL nutné vyšetření kostní dřeně či mízní uzliny. Vyšetření kostní dřeně je zpravidla prováděno k objasnění etiologie anémie či trombocytopenie – odlišení infiltrace při CLL, autoimunitní příčiny apod. Exstirpace a histologické vyšetření mízní uzliny je indikováno v případě, kdy není diagnóza CLL jednoznačná (např. netypický imunofenotyp) či při podezření na transformaci do jiné lymfoproliferace (Richterův syndrom) [1].
![Diagnostická kritéria CLL podle International Workshop on CLL
(IWCLL) [1]](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image_pdf/9fe1441eb8fafc9f5b2e46b3bf39f74c.jpeg)
Buňky CLL mají charakteristický imunofenotyp definovaný koexpresí znaků CD5, CD19 a CD23. K cytometrické diagnóze CLL a diferenciální diagnostice je možno využít skórovací systém Royal Marsden založený na expresi pěti povrchových znaků nádorových lymfocytů – tabulka 2 [3]. Většina případů CLL má vysoké skóre (4–5 bodů), ostatní B-lymfoproliferace mají skóre nízké (0–2 body). Diferenciálně diagnosticky je třeba od CLL odlišit zejména leukemizovaný lymfom z plášťových buněk a B-prolymfocytární leukemii. K odlišení CLL od ostatních lymfoidních malignit může dobře posloužit znak CD200, který je u CLL silně exprimován, a dále znaky CD43, CD79b, CD81, CD10 a ROR1 [4].

Lymfom z malých lymfocytů (small lymphocytic lymphoma, SLL) má identický imunofenotyp jako CLL; nejzásadnějším rozdílem vůči CLL je nesplnění kritéria periferní lymfocytózy. Aktuální klasifikace Světové zdravotnické organizace uvádí CLL a SLL jako společnou jednotku CLL/SLL. Diagnóza SLL by v případě nepřítomnosti klonu v periferní krvi či kostní dřeni měla být potvrzena histologickým vyšetřením mízní uzliny, pokud je to možné [1]. Pokud je v rámci SLL přítomna cytopenie způsobená infiltrací kostní dřeně, mělo by onemocnění být podle nových doporučení IWCLL 2018 považováno za CLL bez ohledu na počet lymfocytů v periferní krvi [1].
Nález klonální B lymfocytózy < 5 x 109/l s imunofenotypem typickým pro CLL, ale bez současné lymfadenomegalie a organomegalie, cytopenie nebo B-příznaků definuje monoklonální B lymfocytózu (MBL), která je prekancerózou CLL. V 1–2 % případů ročně může MBL progredovat do CLL [5].
2. Doporučená vyšetření u nemocných s novědiagnostikovanou CLL
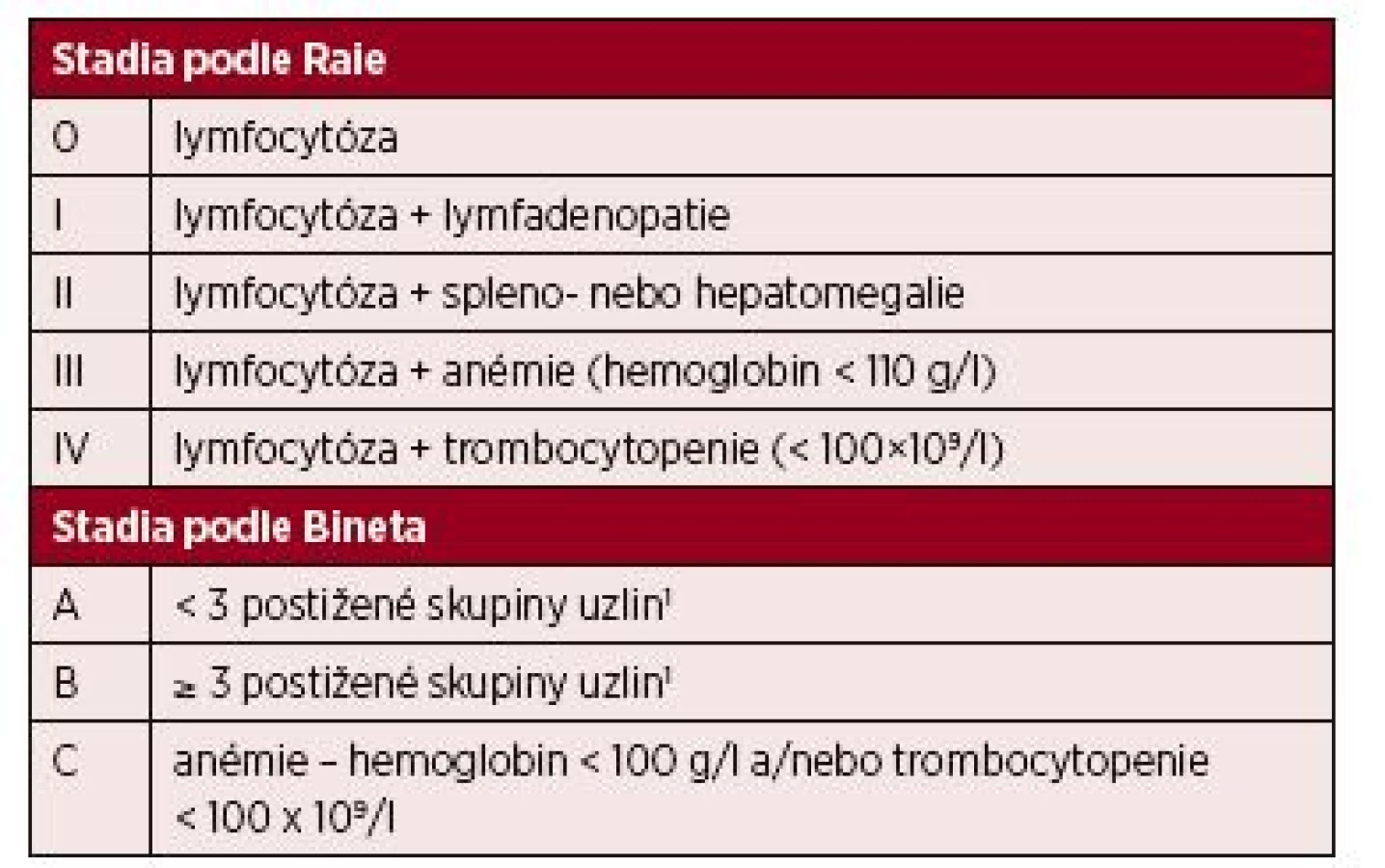
Při diagnóze onemocnění se stanovuje na základě fyzikálního vyšetření a vyšetření krevního obrazu klinické stadium podle Raie nebo Bineta – tabulka 3 [6, 7]. Další vyšetření doporučená v době diagnózy CLL shrnuje tabulka 4 [8].
![Minimální doporučený rozsah vyšetření u nemocných s nově
zjištěnou CLL [7]](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image_pdf/7875f9699ee90b5cc7d48032723cf73e.jpeg)
3. Prognostická vyšetření u CLL
Vzhledem k mimořádné klinické variabilitě nemocných s CLL, a to i v rámci jednoho klinického stadia, je doporučeno u nemocných, kteří jsou kandidáty intenzivní/cílené léčby, zvážit vyšetření umožňující individuální prognózu zpřesnit. Mezi ně patří zejména: vyšetření genetických aberací fluorescenční in situ hybridizací (FISH – delece 13q, 11q, 17p, trizomie 12) [9], stanovení mutačního stavu genů pro variabilní část těžkého řetězce imunoglobulinu (IGHV) [10] a vyšetření mutací genu TP53 [11, 12]. Tyto prognostické faktory mají zásadní vliv na délku období bez léčby, období do progrese a celkové přežití. V současné době je proto u nemocných léčených protokoly ovlivňujícími přirozený průběh CLL (kombinované fludarabinové či bendamustinové režimy, inhibitory signálních drah B-buněčného receptoru [BCR], alemtuzumab či alogenní transplantace) doporučeno provést vyšetření chromozomálních abnormalit pomocí FISH, zejména deleci 17p a analýzu mutací TP53 před zahájením 1. linie léčby a také před každou novou linií léčby [8, 13]. Význam nově objevených mutací genů BIRC3, NOTCH1, MYD88, SF3B1 a dalších, je v současné době intenzivně zkoumán; vyšetření těchto mutací není v současné době vyžadováno pro běžnou praxi [8]. Stejně tak je diskutována role minoritních klonů s mutacemi TP53 pro prognózu onemocnění [11]. V posledních letech byla publikována data poukazující na negativní prognostický a prediktivní význam komplexního karyotypu (definovaného jako 3 a více chromozomových změn) [14–16]. I když zatím nemáme in extenso publikovaná prospektivní data, je vhodné u nemocných s CLL provádět kromě FISH rovněž vyšetření karyotypu periferní krve po stimulaci mitogeny (např. CpG oligonukleotidy a interleukinem-2) a výsledky vyhodnotit podle doporučení a platné cytogenetické nomenklatury [16, 17].
Zobrazovací vyšetření u CLL nabývají stále na větším významu vzhledem k častému výskytu nitrobřišní lymfadenopatie a léčbě inhibitory BCR/Bcl-2. Je proto vhodné v rámci stanovení rozsahu onemocnění před léčbou a při hodnocení léčebné odpovědi provádět přinejmenším ultrasonografii břicha a RTG hrudníku, u mladších nemocných pak CT hrudníku a břicha [1, 8].
4. Léčba CLL
Léčba nemocných s CLL mimo klinické studie je individuální s přihlédnutím ke všem okolnostem aktuálního stavu nemocného, přidruženým onemocněním, přáním apod. Vzhledem k tomu, že pouze klinický výzkum vede ke zlepšování výsledků léčby, měla by být každému vhodnému nemocnému s CLL nabídnuta účast v některé z klinických studií probíhajících v České republice. Zásadní je dobrá komunikace mezi regionálními hematologickými pracovišti a centry vysoce specializované hematoonkologické péče pro dospělé. Velice důležité je do těchto center včas odeslat mladší nemocné s vysoce nepříznivým průběhem CLL ke zvážení alogenní transplantace krvetvorných kmenových buněk.
4. 1 Indikace k léčbě CLL
Řídíme se podle mezinárodně uznávaných kritérií IWCLL 2018 [1]. Nemocní s pokročilým onemocněním (stadium III a IV podle Raie nebo stadium C podle Bineta) jsou indikováni k zahájení léčby. Pouze u malé skupiny nemocných se stabilní mírnou anémií či trombocytopenií je možno léčbu nezahajovat a pečlivě nemocného monitorovat. U nemocných se středně pokročilým onemocněním (Rai I/II či Binet B) by měla být léčba zahájena pouze při průkazu aktivity CLL [1, 8].
K definici aktivního onemocnění a zahájení léčby je nutné splnit nejméně jedno z následujících kritérií:
- Průkaz progresivního selhání kostní dřeně, které se projevuje rozvojem nebo zhoršením anémie a/nebo trombocytopenie.
- Masivní (tj. > 6 cm pod levým žeberním obloukem) nebo progresivní nebo symptomatická splenomegalie.
- Masivní lymfadenopatie (tj. > 10 cm v nejdelším průměru) nebo progresivní nebo symptomatická lymfadenopatie.
- Progresivní lymfocytóza se vzestupem > 50 % během 2 měsíců nebo doba zdvojnásobení počtu lymfocytů (LDT) kratší než 6 měsíců. Zejména u nemocných s úvodní lymfocytózou pod 50 x 109/l je třeba vyloučit jiné faktory, které mohou přispívat k lymfocytóze nebo lymfadenopatii, např. infekce. Progrese lymfocytózy se může u některých nemocných zpomalit, proto je vhodné zejména při absenci ostatních známek aktivity CLL pečlivě sledovat klinický vývoj.
- Autoimunitní anémie a/nebo trombocytopenie špatně odpovídající na kortikosteroidy nebo jinou standardní léčbu.
- Nejméně jeden z následujících systémových příznaků souvisejících s onemocněním:
- a) nechtěný úbytek hmotnosti ≥ 10 % v průběhu předchozích 6 měsíců,
- b) významná únava (tj. výkonnostní stav podle ECOG 2 nebo horší, nemožnost pracovat nebo provádět obvyklé činnosti),
- c) horečky nad 38 °C po dobu 2 nebo více týdnů bez průkazu infekce,
- d) noční pocení po dobu delší než 1 měsíc bez průkazu infekce.
Autoimunní hemolytická anémie a autoimunní trombocytopenie nejsou indikací k cytoredukční léčbě, nejsou-li současně splněna jiná kritéria aktivity CLL. Nemocní s autoimunními cytopeniemi by měli být léčeni imunosupresivní léčbou, např. kortikoterapií. Hypogamaglobulinemie nebo monoklonální/oligoklonální paraproteinemie není sama o sobě důvodem zahájení léčby. U nemocných s CLL může být značně zvýšený počet leukocytů, avšak u CLL jsou příznaky z leukostázy velmi vzácné. Absolutní počet leukocytů by proto neměl být používán jako samostatný indikátor léčby u asymptomatických nemocných. Samotná přítomnost nepříznivých prognostických faktorů (nemutované IGHV geny, delece 17p apod.) bez klinické aktivity CLL není indikací k zahájení léčby [1, 8].
4. 2 Vyšetření před léčbou
Před zahájením léčby je vhodné provést tato vyšetření:
- Stanovení rozsahu onemocnění: fyzikální vyšetření a ultrasonografie břicha + rentgenový snímek hrudníku, u mladších nemocných eventuálně CT hrudníku a břicha. Vyšetření kostní dřeně je nutné zejména u nemocných s anémií či trombocytopenií k posouzení etiologie. Dále je doporučeno provést vyšetření FISH, zejména delece 17p, a mutační analýzu TP53, jejichž přítomnost může ovlivnit volbu léčby [1, 8].
- Vyšetření nutná ke stanovení bezpečnosti léčby: kromě základních biochemických ukazatelů zejména Coombsův (antiglobulinový) test + ukazatele hemolýzy (bilirubin, laktátdehydrogenáza, retikulocyty, haptoglobin) [8]. U nemocných s aktivní autoimunitní hemolýzou není zpravidla použití fludarabinu doporučováno. Samotná pozitivita Coombsova testu bez aktivní hemolýzy však není důvodem fludarabin nepoužít. Dále vyšetřujeme sérologie virových hepatitid k vyloučení chronické hepatitidy B a C (HBsAg, anti-HBs, anti-HBc total a IgM, anti-HCV); u mladších nemocných je vhodné vyšetření sérologie HIV, před intenzivní léčbou vyšetření sérologie cytomegaloviru (CMV) [8]. Velmi důležité je stanovení funkce ledvin vzhledem k renálnímu vylučování řady cytostatik, např. fludarabinu a cyklofosfamidu. Lze použít např. vypočtenou clearance kreatininu podle Cockcroftovy a Gaultovy rovnice [18].
- Posouzení celkového stavu a přidružených onemocnění je velice důležité pro stanovení cílů léčby [19]. Významná část nemocných s CLL má již v době diagnózy významná přidružená onemocnění [19], jejichž počet či závažnost ovlivňují celkové přežití [20–22]. Důležitější než kalendářní je biologický věk. Stanovujeme výkonnostní stav podle ECOG, vhodné je však také posoudit počet a závažnost přidružených onemocnění. Je možno využít např. systém Cumulative Illness Rating Scale (CIRS) [22–24]. Základním způsobem lze rozdělit nemocné do tří skupin:
- a) nemocní v dobrém stavu bez závažných přidružených onemocnění („Go-Go“). V klinických studiích je používána hranice CIRS ≤ 6 bodů k definici této skupiny;
- b) nemocní s významnými přidruženými onemocněními, ale v uspokojivém stavu („Slow-Go“);
- c) nemocní ve špatném stavu s těžkými komorbiditami („No-Go“) [19].
Ke stanovení intenzity a typu léčby je v současné době nejdůležitější pečlivé individuální posouzení nemocného.
4. Zásadní je také vzít v úvahu přání nemocného. Cíle a představy lékaře a nemocného se nemusí vždy shodovat, proto je důležité s nemocným prodiskutovat všechny vhodné možnosti léčby a podrobně vysvětlit výhody a nevýhody jednotlivých postupů.
4. 3 Hodnocení léčebné odpovědi
Řídí se doporučeními IWCLL [1]. Hodnocení léčebné odpovědi je vhodné provést po 2–3 měsících od ukončení léčby z důvodu umožnění regenerace v krevním obraze po myelotoxických režimech typu FCR. V případě léčby inhibitory BCR a Bcl-2, kdy je léčba dlouhodobá, je zapotřebí opakované posouzení léčebné odpovědi během léčby. Hodnocení léčebné odpovědi se opírá o fyzikální vyšetření a vyšetření krevního obrazu s diferenciálním rozpočtem leukocytů. Tato vyšetření mohou být doplněna o vyšetření kostní dřeně a vyšetření zobrazovacími metodami. Ideálním cílem léčby je dosažení kompletní remise (CR). Jednotlivé kategorie léčebné odpovědi a jejich definice jsou shrnuty v tabulce 5. Vzhledem k tomu, že nové preparáty zasahující do signalizace B-buněčného receptoru (ibrutinib, idelalisib) způsobují redistribuci maligních lymfocytů z mízních uzlin do periferní krve s následnou lymfocytózou, která může být přechodná v úvodu léčby, ale také může trvat i řadu měsíců, byla navržena nová kategorie „částečná odpověď s lymfocytózou“ (PR-L) [25]. Jde o případ, kdy nemocný nesplní kritérium PR z důvodu lymfocytózy vyvolané léčbou. Tato kategorie slouží zejména k tomu, aby nemocným v léčbě inhibitory BCR nebyla předčasně ukončena léčba; PR s lymfocytózou by neměla být považována za progresi CLL [25].
![Hodnocení léčebné odpovědi u CLL, upraveno podle [1]](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image_pdf/3ac3c680b85dc9b16045eb0f302ad375.jpeg)
Definice kompletní remise (CR): splněna veškerá kritéria skupiny A i B a nemocný nesmí mít systémové příznaky spojené s CLL. Vyšetření kostní dřeně není nezbytně nutné pro hodnocení kompletní remise mimo klinické studie.
Definice parciální remise (PR): splněna nejméně 2 kritéria ze skupiny A + nejméně jedno kritérium ze skupiny B.
Definice stabilní choroby (SD): nedosažení léčebné odpovědi (tedy CR či PR), zároveň nejsou splněna kritéria progresivní choroby.
Definice progresivní choroby (PD): splněno nejméně jedno z kritérií skupiny A či B.
Pro splnění definice CR a PR musí být hodnocené parametry stabilní nejméně 2 měsíce.
Kompletní remise s neúplnou restitucí krevního obrazu (CRi) je definována stejně jako CR, je však přítomna reziduální cytopenie po léčbě (snížený ANC a/nebo hemoglobin a/nebo trombocyty).
Pokud bylo dosaženo PR na základě hodnocení krevního obrazu a organomegalie, je vyšetření kostní dřeně irelevantní, neboť nezmění kategorii léčebné odpovědi.
Do kategorie „progresivní choroba“ se řadí také transformace do lymfoproliferace vyšší malignity (Richterův syndrom).
Relaps je stav, kdy u nemocného, jenž dosáhl po léčbě CR nebo PR, dojde za ≥ 6 měsíců od skončení léčby k progresi nemoci (viz definice PD). Refrakterní onemocnění je definováno jako nedosažení CR nebo PR či relaps/progrese do 6 měsíců od ukončení léčby.
4. 4 Léčba 1. linie
4. 4. 1 Léčba 1. linie u nemocných bez významných přidružených onemocnění
Na základě výsledků randomizované studie CLL8 je režim FCR (fludarabin, cyklofosfamid, rituximab) považován za hlavní léčebnou možnost u nemocných v dobrém stavu bez závažných přidružených onemocnění a s normální funkcí ledvin [8, 26–28] – tabulka 6. V případě delece 17p/mutace TP53 a nevhodnosti léčby chemoimunoterapií je vhodné použít ibrutinib [8, 29]. Idelalisib v kombinaci s rituximabem je možno použít u nemocných s delecí či mutací TP53 při nevhodnosti jakékoliv jiné léčby [30]. Vzhledem ke klinické variabilitě u nemocných s CLL je často nutno léčbu individualizovat.

**v případě nevhodnosti jakékoliv jiné léčby.
FCR = fludarabin + cyklofosfamid + rituximab; BR = bendamustin + rituximab; BO = bendamustin + ofatumumab; RCD = rituximab + cyklofosfamid + dexametazon
Další léčebné možnosti (řazeno abecedně):
- Alemtuzumab [31] (Pozn. Alemtuzumab není pro léčbu CLL registrován, je dostupný v ČR v rámci specifického léčebného programu.)
- Bendamustin + rituximab (BR) jako alternativa k režimu FCR u nemocných ≥ 65 let věku, zejména v případě vysokého rizika infekcí či předchozích závažných infekcí [27, 32]
- Rituximab, cyklofosfamid, dexametazon (RCD) či obdobné režimy s vysokodávkovanými kortikoidy [28, 33].
4. 4. 2 Léčba 1. linie u nemocných s významnými přidruženými chorobami
U nemocných s významnými komorbiditami (např. skóre komorbidit CIRS > 6) či clearance kreatininu < 70 ml/min., kteří tedy nejsou vhodní k léčbě plnodávkovaným protokolem FCR, jsou k dispozici tyto hlavní možnosti (v abecedním pořadí):
- Obinutuzumab + chlorambucil [34, 35]
- Ofatumumab + chlorambucil [36]
- Rituximab + bendamustin (BR) [37]
- Rituximab + chlorambucil [35, 37, 38].
Podle SPC přípravku je v léčbě první linie u pacientů bez defektu TP53 možno použít rovněž ibrutinib [39]. ČSCLL však doporučuje v současné době ponechat tento preparát až pro případy léčby pacientů s relapsem onemocnění. Chybí nám navíc přímé porovnání mezi monoterapií ibrutinibem a současným standardem pro komorbidní nemocné (chlorambucil + anti-CD20 protilátka). Navíc léčba chlorambucilem + anti-CD20 protilátkou je časově omezená ve srovnání s podáváním ibrutinibu, jenž je užíván do progrese či závažné toxicity.
U nemocných s delecí 17p/mutací TP53 nevhodných k léčbě chemoimunoterapií je vhodné použít ibrutinib [8]. Idelalisib v kombinaci s rituximabem je možno použít u nemocných s delecí či mutací TP53 při nevhodnosti jakékoliv jiné léčby [30].
Další možnosti léčby (v abecedním pořadí):
- Alemtuzumab [31] (Pozn. Alemtuzumab není pro léčbu CLL registrován, je dostupný v ČR v rámci specifického léčebného programu.)
- Bendamustin + ofatumumab (BO) [40]
- FCR se sníženými dávkami chemoterapie (low-dose FCR) [41]
- Rituximab, cyklofosfamid, dexametazon (RCD) či obdobné režimy s vysokodávkovanými kortikoidy [33, 42, 43].
U těžce komorbidních nemocných, u kterých lze očekávat krátké přežití z důvodu přidružených onemocnění, je cílem léčby ovlivnění symptomů CLL s minimem nežádoucích účinků. Lze využít např. chlorambucil v monoterapii, nízkodávkovaný cyklofosfamid v monoterapii či kortikoterapii.
4. 5 Léčba relapsu/refrakterní CLL
Volba vhodného léčebného režimu je dána celkovým stavem a věkem pacienta a také odpovědí na předchozí léčbu a délkou jejího trvání. Pacienti s pozdním relapsem po chemoimunoterapii, u nichž léčebná odpověď trvala déle než 2–3 roky, mohou profitovat z opakované aplikace chemoimunoterapie [8]. V případě časných relapsů (do 2–3 let), refrakterní choroby či přítomnosti delece 17p/mutace TP53 je nutné změnit léčebný přístup a použít cílenou léčbu inhibitory BCR či Bcl-2 [8].
4. 5. 1 Léčba relapsu/refrakterní CLL u nemocných bez významných přidružených onemocnění
Hlavní léčebné možnosti (řazené podle abecedy) představují ibrutinib a idelalisib zasahující do signalizace BCR [8, 44, 45] – tabulka 7. Tyto preparáty mají zpravidla mírnou hematologickou toxicitu, vyskytuje se však odlišný profil nežádoucích účinků, které je zapotřebí pečlivě sledovat (průjem/kolitida, zvýšení jaterních testů, pneumonitida, krvácení, kožní exantém apod.) [44–46]. Inhibitor Bcl-2 venetoklax je indikován:
- a) pro léčbu nemocných s delecí 17p či mutací TP53 po selhání či při nevhodnosti léčby inhibitorem BCR;
- b) pro léčbu nemocných bez delece či mutace TP53 po selhání chemoimunoterapie a léčby BCR inhibitorem.
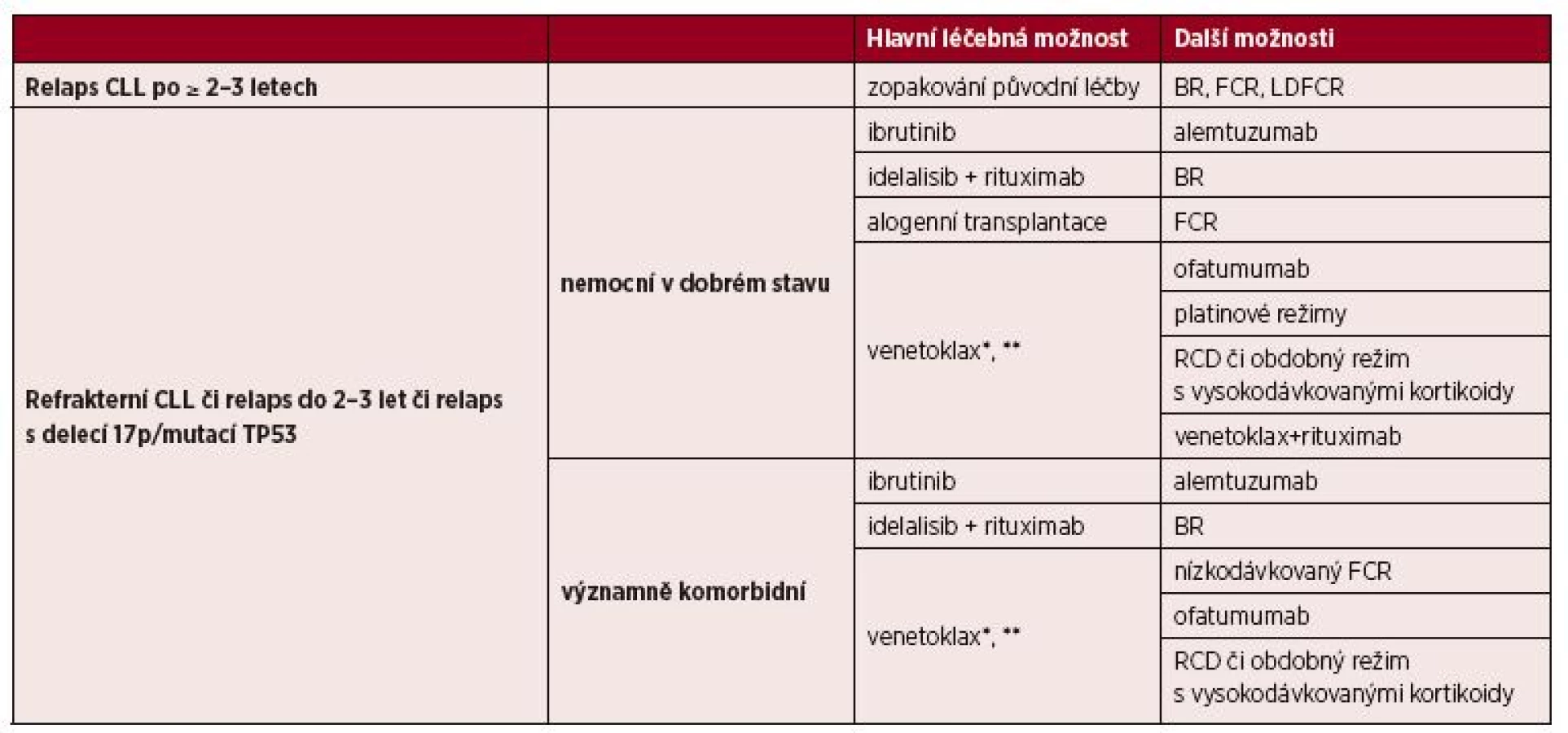
**U nemocných bez delece či mutace TP53 po selhání chemoimunoterapie i léčby BCR inhibitorem.
BR = bendamustin + rituximab; FCR = fludarabin + cyklofosfamid + rituximab; LDFCR – nízkodávkovaný FCR; RCD = rituximab + cyklofosfamid + dexametazon
Venetoklax tedy představuje hlavní možnost po selhání léčby ibrutinibem či idelalisibem [47, 48]. Zcela nově byly publikovány výsledky randomizované studie u nemocných s relapsem či refrakterní CLL, ve které kombinace venetoklaxu s rituximabem (VR) dosáhla významně lepších výsledků než režim BR [49]. Není však dosud jasné, kde bude místo tohoto režimu v rámci léčebných algoritmů, neboť nemocní léčení kombinací VR byli výrazně méně předléčeni než nemocní ve studiích s monoterapií venetoklaxem.
Ve dvou randomizovaných, dvojitě slepých studiích byl prokázán zřetelný přínos pro nemocné s kombinací režimu BR a inhibitoru BCR ibrutinibu [50] či BR + idelalisibu [51]. Je však otázkou, jaký význam v těchto režimech hrála přítomnost bendamustinu a zda tyto protokoly najdou širší uplatnění v nastávající éře léčby bez chemoterapie.
Další možnosti léčby (řazení podle abecedy):
- alemtuzumab [52, 53]
- BR (bendamustin + rituximab) [54, 55] (Pozn. Bendamustin v léčbě relapsu/refrakterní CLL nemá indikaci podle souhrnu údajů o přípravku („off-label“ indikace), nutno žádat o úhradu revizního lékaře.
- FCR [56–58]
- ofatumumab u nemocných refrakterních na fluda-rabin i alemtuzumab [59]
- RCD (rituximab, cyklofosfamid, dexametazon) či obdobné režimy s vysokodávkovanými kortikoidy, zejména u refrakterních nemocných [33, 42, 43]
- kombinace rituximabu s intenzivními platinovými režimy (např. R-DHAP) u refrakterních nemocných mladšího věku ve velmi dobrém stavu bez závažných komorbidit [60].
4. 5. 2 Léčba relapsu/refrakterní CLL u nemocných s významnými přidruženými onemocněními
Hlavní možnosti (řazeno podle abecedy):
- ibrutinib [44, 61, 62]
- •delalisib + rituximab [45]
- venetoklax [47, 48].
Další možnosti (řazeno podle abecedy):
- alemtuzumab [52, 53]
- BR (bendamustin + rituximab) [54, 55] (Pozn. Bendamustin v léčbě relapsu/refrakterní CLL ne-má indikaci podle souhrnu údajů o přípravku („off-label“ indikace), nutno žádat o úhradu revizního lékaře.
- FCR v redukovaných dávkách („low dose“ FCR) [41]
- ofatumumab u nemocných refrakterních na fludarabin i alemtuzumab [59], RCD (rituximab, cyklofosfamid, dexametazon) či obdobné režimy s vysokodávkovanými kortikoidy [33, 42, 43].
U relabujících nemocných s nepříznivou genetikou (delece 17p/mutace TP53) by mělo být i v případě pozdního relapsu zvažováno podání ibrutinibu či idelalisibu, které nabízejí lepší kontrolu nemoci v porovnání s ostatní dosud dostupnou léčbou, s výjimkou alogenní transplantace krvetvorných kmenových buněk. U nemocných po selhání inhibitoru BCR je nutno zvažovat podání venetoklaxu.
Nemocní s refrakterní CLL by měli být včas konzultováni v některém z center vysoce specializované hematoonkologické péče pro dospělé, které by mělo řídit další strategii léčby, indikovat případně léčbu moderními molekulami či monoklonálními protilátkami, ideálně v rámci klinických studií, a také zvážit provedení alogenní transplantace krvetvorných buněk.
4. 6 Minimální reziduální nemoc
Moderní léčebné protokoly vedou u významné části nemocných ke snížení leukemického klonu pod úroveň detekce běžnými metodami; zbytkové leukemické elementy (minimální reziduální nemoc, MRN) mohou být zjistitelné pouze velmi citlivými metodami. V literatuře přibývá důkazů o prodloužení přežití bez progrese a celkového přežití u nemocných, u kterých bylo dosaženo negativity MRN [63]. Prognostický dopad MRN navíc není závislý na podané léčbě či na jiných rizikových faktorech (mutační stav IGHV, chromozomové aberace apod.). Vyšetření MRN v periferní krvi či kostní dřeni metodami s citlivostí minimálně 10-4 (pomocí 4–8 barevné průtokové cytometrie, PCR s individuálně připravenými primery či pomocí sekvenování nové generace) je vhodné zvážit u nemocných po intenzivní léčbě (např. kombinované fludarabinové režimy, alemtuzumab, režimy s novými monoklonálními protilátkami, léčba inhibitorem Bcl-2, alogenní transplantace) [64]. Dosud se nejedná o vyšetření povinné [8], ale význam analýz MRN v budoucnu pravděpodobně významně vzroste s tím, jak bude pomocí nových léčebných kombinací narůstat počet dosažených negativit MRN. Vhodné je rovněž u pacientů po alogenní transplantaci krvetvorných buněk.
4. 7 Transplantace krvetvorných buněk
Alogenní transplantace krvetvorných buněk by měla být včas zvažována u každého nemocného s vysoce rizikovou CLL (refrakterní na purinová analoga, relaps < 24–36 měsíců po chemoimunoterapii, delece 17p/mutace TP53), respektive obecně u nemocných s nepříznivým klinickým průběhem [65, 66]. Tito nemocní by měli být včas konzultováni v některém z center vysoce specializované hematoonkologické péče pro dospělé vzhledem k tomu, že transplantace by měla být provedena včas, dříve, než je onemocnění kompletně refrakterní a pacient masivně předléčený.
Alogenní transplantace má u CLL kurativní potenciál a nabízí dlouhodobou kontrolu nemoci. Část nemocných dosahuje negativity na úrovni minimální reziduální nemoci a výsledek transplantace není nepříznivě ovlivněn negativními genetickými rizikovými faktory (delece 17p/mutace TP53). Transplantace jeovšem zatížena cca 15–30% mortalitou v důsledku infekcí nebo toxicity v prvních dvou letech a až u 25 % pacientů omezuje jejich kvalitu života chronická reakce štěpu proti hostiteli (GVHD) [66].
Nové molekuly (inhibitory BCR/Bcl-2) změnily doporučení pro indikaci a zejména pro načasování alogenní transplantace. Transplantaci není v současné době nutné provádět u CLL s aberací TP53 v první remisi, s ohledem na to, že je velká šance na dosažení druhé remise novými molekulami. Indikace se více přesouvá do období relapsu onemocnění a kandidáty transplantace se stávají nemocní s relabující/refrakterní CLL a nedostatečnou odpovědí na moderní léky [66]. Přímé porovnání alogenní transplantace s novými léky (inhibitory BCR či Bcl-2) však zatím nemáme a nevíme ani, jaká bude efektivita transplantace při progresi po inhibitorech. Pacientům s vysoce rizikovou CLL by měla být v současné době nabídnuta některá z nových molekul (ibrutinib, idelalisib, venetoklax, případně další)[8]. Po dosažení odpovědi může být zvažována konsolidace alogenní transplantací nebo pokračující léčba novými molekulami do progrese onemocnění. Transplantační přístup bude upřednostňován u relabující/refrakterní CLL s delecí 17p/mutací TP53, a to především u mladších pacientů s dobře shodným dárcem [66]. Netransplantační cesta je naopak vhodnější pro starší nemocné s komorbiditami, zejména při absenci nepříznivé genetiky a nedostupnosti vhodného dárce.
Pro alogenní transplantaci se v současné době volí nejčastěji použití nemyeloablativního přípravného režimu, který přináší nižší peritransplantační mortalitu a možnost provést transplantaci u nemocných do 65 (výjimečně 70) let. Tato metoda je v současné praxi upřednostňována před klasickým myeloablativním přístupem [66].
4. 8 Transformace CLL (Richterův syndrom, RS)
Transformace CLL do jiné lymfoproliferace, nejčastěji difuzního velkobuněčného lymfomu (DLBCL), vzácně Hodgkinova lymfomu, je prognosticky vysoce nepříznivý jev. K transformaci dochází u 2–10 % pacientů v průběhu jejich onemocnění, s pravděpodobností 0,5–1 % za rok [67–69]. Na transformaci CLL je nutno pomýšlet při následujících nálezech: významné zvýšení LDH, progrese lymfadenopatie, zejména asymetrické v jedné oblasti, rozvoj B-příznaků, progrese při terapii. Při podezření na Richterův syndrom je možné využít vyšetření PET/CT. K potvrzení Richterovy transformace je rozhodující exstirpace mízní uzliny s histologickým vyšetřením [68]. U nemocných s Richterovým syndromem je indikována léčba pro agresivní lymfomy, tzn. chemoimunoterapie využívající rituximab (R-CHOP a další režimy), včetně léčby transplantační. Hlavním faktorem, který určuje prognózu pacienta s RS, je klonální příslušnost DLBCL k přítomné CLL. Na základě sekvenování IGHV genu lze zjistit, že kolem 20 % případů transformace představuje nově vzniklý DLBCL bez klonálního vztahu k CLL [68]. Prognóza tohoto RS je stejná jako u de novo vzniklého DLBCL; léčí se proto stejným způsobem, nejčastěji protokolem R-CHOP. Při nedosažení kompletní remise je indikována záchranná léčba následovaná autologní transplantací. V 80 % případů Richterovy transformace do DLBCL jde o onemocnění vzniklé klonálně z původní CLL s výrazně nepříznivou prognózou (medián přežití 8–14 měsíců). Jako úvodní léčba je opět vhodný režim R-CHOP a po chemoimunoterapii se zvažuje transplantační léčba. Alogenní transplantace je preferována u mladších nemocných v dobrém celkovém stavu a dostupným vhodným dárcem. Autologní transplantace pro Richterův syndrom se volí u nemocných, kteří nejsou kandidáty alogenní transplantace z důvodu věku či komorbidit. V případě transformace do Hodgkinova lymfomu se použije chemoterapie určená k léčbě tohoto onemocnění (např. ABVD) [68].
4.9 Podpůrná léčba
Nemocní s CLL mají vysoké riziko infekčních komplikací, které souvisí jak s defekty imunity v důsledku působení choroby samotné, tak i v důsledku imunosuprese navozené léčbou. Podpůrná léčba je tedy cílena zejména na prevenci a včasnou léčbu širokého spektra infekcí včetně oportunních nákaz. U každého nemocného léčeného protokoly obsahujícími fludarabin či kortikoidy by měla být zvážena protiinfekční profylaxe (sulfametoxazol + trimetoprim, antivirotika, eventuálně antimykotika). Prevence pneumocystové pneumonie pomocí kombinace sulfametoxazol + trimetoprim či vhodné alternativy je nutná u všech nemocných na léčbě idelalisibem [30]. Kombinace sulfametoxazol/trimetoprim + antivirotika je povinná u protokolů obsahujících alemtuzumab či vysokodávkované kortikoidy [70, 71]. Při léčbě alemtuzumabem či idelalisibem je dále nutné pravidelné klinické a laboratorní monitorování CMV reaktivace a v případě klinicky významné reaktivace CMV pak přerušení terapie a zahájení antivirotické léčby (ganciclovir, valganciklovir) [70, 71].
Podpůrná terapie při léčbě venetoklaxem: použití venetoklaxu bylo v časných klinických studiích spojenu s rozvojem syndromu z rozpadu nádoru (tumor lysis syndrome, TLS). Je proto nutné dodržet dávkovací schéma podle doporučení pro venetoklax [72], podle kterých je první dávka 20 mg a postupně se dávka zvyšuje v týdenních intervalech na 50, 100, 200 až na cílových 400 mg denně. Během tohoto období je nutné pečlivé monitorování biochemických parametrů zaměřených na známky TLS a vyšetřování krevního obrazu. Před zahájením léčby venetoklaxem by měl být nemocný masivně hydratován, podán allopurinol, případně rasburikáza). Doporučeno je zahájení léčby venetoklaxem za hospitalizace, zejména u nemocných s vysokou nádorovou náloží [67].
U nemocných s opakovanými bakteriálními infekcemi a sníženou sérovou koncentrací IgG pod 5 g/l by měla být vedle antimikrobiální profylaxe dále individuálně zvážena i substituce parenterálními imunoglobuliny [8, 73, 74]. U pacientů s CLL je dále doporučováno i každoroční očkování proti chřipce a pravidelné očkování proti pneumokokům (každých 5 let, upřednostňována je konjugovaná vakcína) [75]. Je nutné mít na paměti, že pacienti s CLL mají nižší odpověď na očkování a v době chřipkové sezony se doporučuje zvýšená pozornost i u těch nemocných, kteří očkování podstoupili. Pacienti léčení rituximabem by měli být očkováni až poté, co dojde k regeneraci B lymfocytů. Pacientům s CLL se nesmí podávat živé vakcíny. Imunosupresivní terapie může vést také k reaktivaci hepatitidy B nebo C, proto by pacienti před léčbou CLL měli být vyšetřeni na hepatitidy B a C (HBsAg, anti-HBs, anti-HBc total a IgM, anti-HCV) [8] a v případě průkazu proběhlé infekce je vhodné pacienta dále konzultovat s hepatologem či infektologem, v některých případech je nutná antivirotická profylaxe nebo terapie. U pacientů s CLL léčených chemoterapií, imunoterapií či imunochemoterapií, u kterých dojde k rozvoji anémie, je vhodná léčba erytropoézu-stimulujícími proteiny (erytropoetin, darbepoetin) v souladu s mezinárodními doporučeními [76]. U nemocných, kteří jsou těžce imunosuprimováni, zejména po léčbě fludarabinem či alemtuzumabem a po alogenní transplantaci, stejně jako u nemocných, u kterých se uvažuje o provedení alogenní transplantace v budoucnu, by měly být používány ozářené transfuzní přípravky vzhledem ke zvýšenému riziku rozvoje reakce štěpu proti hostiteli spojené s transfuzí [73]. Vzhledem k tomu, že intenzivní léčebné protokoly, zejména protokoly obsahující fludarabin či alemtuzumab, jsou spojeny s vysokým rizikem febrilní neutropenie, je vhodné na základě individuálního rizika zvážit primární profylaxi febrilní neutropenie pomocí granulocytového kolonie stimulujícího faktoru (G-CSF) v souladu s doporučeními mezinárodních společností [77]. V případě výskytu febrilní neutropenie při léčbě je vhodné podat G-CSF v dalších cyklech léčby jako sekundární profylaxi febrilní neutropenie.
Nemocní s CLL mají 2–5krát vyšší riziko vzniku sekundárních malignit. Časté jsou zejména kožní nádory, dále karcinom prostaty, plicní a kolorektální karcinom [78]. Je proto vhodné myslet u těchto nemocných ve spolupráci s praktickým lékařem na pravidelný onkologický screening:
- vyšetření stolice na okultní krvácení,
- kontroly PSA u mužů,
- gynekologické vyšetření a mamografie u žen a
- dermatologické vyšetření [73, 78].
Upozornění: Tato doporučení jsou pouze návodem, jak je možno u nemocných s CLL postupovat. Autoři nenesou žádnou právní zodpovědnost za obsah těchto doporučení ani volbu konkrétního postupu u konkrétního nemocného – ta je plně zodpovědností ošetřujícího lékaře.
Seznam použitých zkratek
ABVD – doxorubicin, bleomycin, vinblastin, dakarbazin
ANC – absolutní počet neutrofilů
ALC – absolutní počet lymfocytů
B2M – beta 2-mikroglobulin
Bcl-2 – B-cell lymphoma 2 gen
BCR – B-buněčný receptor
BO – bendamustin + ofatumumab
BR – bendamustin + rituximab
CIRS – Cumulative Illness Rating Scale
CLL – chronická lymfocytární leukemie
CR – kompletní remise
CT – počítačová tomografie
DLBCL – difuzní B-velkobuněčný lymfom
ECOG – Eastern Cooperative Oncology Group
FCR – fludarabin, cyklofosfamid, rituximab
FISH – fluorescenční in situ hybridizace
G-CSF – granulocytární kolonie stimulující faktor
GVHD – reakce štěpu proti hostiteli
IGHV – variabilní část těžkého řetězce imunoglobulinu
LDH – laktátdehydrogenáza
MBL – monoklonální B-buněčná lymfocytóza
MRN – minimální reziduální nemoc
IWCLL – International Workshop on Chronic Lymphocytic Leukemia
PD – progresivní choroba
PR – parciální remise
PR-L – parciální remise s lymfocytózou
R – rituximab
R-CHOP – rituximab, cyklofosfamid, doxorubicin, vinkristin, prednison
R-DHAP – rituximab, dexametazon, cytosin-arabinosid, cisplatina
RCD – rituximab, cyklofosfamid, dexametazon
RL – revizní lékař
SD – stabilní onemocnění
SLL – lymfom z malých lymfocytů
SPC – Summary of Product Characteristics
TLS – syndrom nádorového rozpadu
TP53 – tumor supresorový gen p53
VR – venetoklax + rituximab
Podíl autorů na přípravě rukopisu
DM, ŠM, SL – podíleli se na napsání manuskriptu, provedli jeho revizi a konečnou úpravu k tisku.
PŠ, JM, PT, UR, ŠM, LD, BM – podíleli se na napsání manuskriptu a provedli jeho revizi.
Prohlášení o spolupráci s farmaceutickýmispolečnostmi
M. Doubek: AbbVie, Angelini, AOP Orphan, Gilead, Janssen-Cilag, Novartis, Roche (honoráře za přednáškovou činnost a konzultace, cestovní granty).
M. Špaček: AbbVie, Gilead, Janssen-Cilag a Roche (honoráře za přednáškovou činnost a konzultace, cestovní granty).
R. Urbanová: Roche (honoráře za přednáškovou činnost, cestovní granty).
M. Šimkovič: Roche, Janssen-Cilag, Gilead (honoráře za přednáškovou činnost, cestovní granty).
D. Lysák: Gilead, Janssen-Cilag, Roche (honoráře za přednáškovou činnost a konzultace, cestovní granty).
L. Smolej: Gilead, Janssen-Cilag, Roche, AbbVie (honoráře za přednáškovou činnost a konzultace, cestovní granty).
M. Brejcha: Gilead, Novartis (honoráře za přednáškovou činnost, cestovní granty).
Poděkování
Tato práce byla podpořena granty IGA MZ ČR NT 13576, IGA MZ ČR NT13493-4/2012, IGA-LF-2015-001, AZV MZ ČR č. 15-30015A-4/2015 a 15-31834A/2015, RVO MZ ČR (FNHK, 00179906) a programem PROGRES Q40/08.
Do redakce doručeno dne 7. 5. 2018.
prof. MUDr. Michael Doubek, Ph.D.
Interní hematologická a onkologická klinika LF MU a FN
Jihlavská 20
625 00 Brno
e-mail: doubek.michael@fnbrno.cz
Zdroje
1. Hallek M, Cheson BD, Catovsky D, et al. Guidelines for diagnosis, indications for treatment, response assessment and supportive management of chronic lymphocytic leukemia. Blood 2018; doi: 10.1182/blood-2017-09-806398.
2. Dearden C. B- and T-cell prolymphocytic leukemia: antibody approaches. Hematology Am Soc Hematol Educ Program 2012:645–651.
3. Moreau EJ, Matutes E, A‘Hern RP, et al. Improvement of the chronic lymphocytic leukemia scoring system with the monoclonal antibody SN8 (CD79b). Am J Clin Pathol 1997;108:378–382.
4. Rawstron AC, Kreuzer KA, Soosapilla A, et al. Reproducible diagnosis of chronic lymphocytic leukemia by flow cytometry: An European Research Initiative on CLL (ERIC) & European Society for Clinical Cell Analysis (ESCCA) Harmonisation project. Cytometry B Clin Cytom 2018;94:121–128.
5. Strati P, Shanafelt TD. Monoclonal B-cell lymphocytosis and early-stage chronic lymphocytic leukemia: diagnosis, natural history, and risk stratification. Blood 2015;126:454–462.
6. Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood 1975;46:219–234.
7. Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981;48:198–206.
8. Eichhorst B, Robak T, Montserrat E, et al. Chronic lymphocytic leukaemia: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2015;26(Suppl 5):v78–v84.
9. Dohner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–1916.
10. Ghia P, Stamatopoulos K, Belessi C, et al. ERIC recommendations on IGHV gene mutational status analysis in chronic lymphocytic leukemia. Leukemia 2007;21:1–3.
11. Malcikova J, Pavlova S, Kozubik KS, Pospisilova S. TP53 mutation analysis in clinical practice: lessons from chronic lymphocytic leukemia. Hum Mutat 2014;35:663–671.
12. Malcikova J, Tausch E, Rossi D, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia-update on methodological approaches and results interpretation. Leukemia 2018;32(5):1070–1080.
13. Pospisilova S, Gonzalez D, Malcikova J, et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia 2012;26:1458–1461.
14. Brejcha M, Stoklasova M, Brychtova Y, et al. Clonal evolution in chronic lymphocytic leukemia detected by fluorescence in situ hybridization and conventional cytogenetics after stimulation with CpG oligonucleotides and interleukin-2: a prospective analysis. Leuk Res 2014;38:170–175.
15. Baliakas P, Iskas M, Gardiner A, et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: a systematic reappraisal of classic cytogenetic data. Am J Hematol 2014;89:249–255.
16. Stevens-Kroef M, Simons A, Rack K, Hastings RJ. Cytogenetic nomenclature and reporting. Methods Mol Biol 2017;1541:303–309.
17. Hastings RJ, Cavani S, Bricarelli FD, Patsalis PC, Kristoffersson U, Co-ordinators EP. Cytogenetic guidelines and quality assurance: a common European framework for quality assessment for constitutional and acquired cytogenetic investigations. Eur J Hum Genet 2007;15:525–527.
18. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron 1976;16:31–41.
19. Hallek M. Chronic lymphocytic leukemia: 2015 Update on diagnosis, risk stratification, and treatment. Am J Hematol 2015;90:446–460.
20. Thurmes P, Call T, Slager S, et al. Comorbid conditions and survival in unselected, newly diagnosed patients with chronic lymphocytic leukemia. Leuk Lymphoma 2008;49:49–56.
21. Extermann M, Overcash J, Lyman GH, Parr J, Balducci L. Comorbidity and functional status are independent in older cancer patients. J Clin Oncol 1998;16:1582–1587.
22. Goede V, Cramer P, Busch R, et al. Interactions between comorbidity and treatment of chronic lymphocytic leukemia: results of German Chronic Lymphocytic Leukemia Study Group trials. Haematologica 2014;99:1095–1100.
23. Parmelee PA, Thuras PD, Katz IR, Lawton MP. Validation of the Cumulative Illness Rating Scale in a geriatric residential population. J Am Geriatr Soc 1995;43:130–137.
24. Salvi F, Miller MD, Grilli A, et al. A manual of guidelines to score the modified cumulative illness rating scale and its validation in acute hospitalized elderly patients. J Am Geriatr Soc 2008;56:1926–1931.
25. Cheson BD, Byrd JC, Rai KR, et al. Novel targeted agents and the need to refine clinical end points in chronic lymphocytic leukemia. J Clin Oncol 2012;30:2820–2822.
26. Cramer P, Langerbeins P, Eichhorst B, Hallek M. Advances in first-line treatment of chronic lymphocytic leukemia current recommendations on management and first-line treatment by the German CLL Study Group (GCLLSG). Eur J Haematol 2016;96:9–18.
27. Hallek M, Fischer K, Fingerle-Rowson G, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet 2010;376:1164–1174.
28. Zelenetz AD, Gordon LI, Wierda WG, et al. Chronic lymphocytic leukemia/small lymphocytic lymphoma, version 1.2015. J Natl Compr Canc Netw 2015;13:326–362.
29. Farooqui MZ, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol 2015;16:169–176.
30. Singh M, Mealing S, Baculea S, Cote S, Whelan J. Impact of novel agents on patient-relevant outcomes in patients with previously untreated chronic lymphocytic leukemia who are not eligible for fludarabine-based therapy. J Med Econ 2017;20:1066–1073.
31. Hillmen P, Skotnicki AB, Robak T, et al. Alemtuzumab compared with chlorambucil as first-line therapy for chronic lymphocytic leukemia. J Clin Oncol 2007;25:5616–5623.
32. Eichhorst B, Fink AM, Bahlo J, et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): an international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol 2016;17:928–942.
33. Michallet AS, Rossignol J, Cazin B, Ysebaert L. Rituximab-cyclophosphamide-dexamethasone combination in management of autoimmune cytopenias associated with chronic lymphocytic leukemia. Leuk Lymphoma 2011;52:1401–1403.
34. Goede V, Fischer K, Busch R, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med 2014;370:1101–1110.
35. Goede V, Fischer K, Engelke A, et al. Obinutuzumab as frontline treatment of chronic lymphocytic leukemia: updated results of the CLL11 study. Leukemia 2015;29:1602–1604.
36. Hillmen P, Robak T, Janssens A, et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): a randomised, multicentre, open-label phase 3 trial. Lancet 2015;385:1873–1883.
37. Michallet AS, Aktan M, Hiddemann W, et al. Rituximab plus bendamustine or chlorambucil for chronic lymphocytic leukemia: primary analysis of the randomized, open-label MABLE study. Haematologica 2018;103:698–706.
38. Hillmen P, Gribben JG, Follows GA, et al. Rituximab plus chlorambucil as first-line treatment for chronic lymphocytic leukemia: Final analysis of an open-label phase II study. J Clin Oncol 2014;32:1236–1241.
39. Sinha R, Redekop WK. Cost-effectiveness of ibrutinib compared with obinutuzumab with chlorambucil in untreated chronic lymphocytic leukemia patients with comorbidities in the United Kingdom. Clin Lymphoma Myeloma Leuk 2018;18:e131–e142.
40. Flinn IW, Panayiotidis P, Afanasyev B, et al. A phase 2, multicenter study investigating ofatumumab and bendamustine combination in patients with untreated or relapsed CLL. Am J Hematol 2016;91:900–906.
41. Smolej L, Brychtova Y, Doubek M, et al. Low-dose FCR is a safeand effective treatment option for elderly/comorbid patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Updated results of project Q-Lite by Czech CLL Study Group. Blood 2014;124:4670.
42. Simkovic M, Motyckova M, Belada D, et al. Five years of experience with rituximab plus high-dose dexamethasone for relapsed/refractory chronic lymphocytic leukemia. Arch Med Sci 2016;12:421–427.
43. Smolej L, Doubek M, Panovska A, et al. Rituximab in combination with high-dose dexamethasone for the treatment of relapsed/refractory chronic lymphocytic leukemia. Leuk Res 2012;36:1278–1282.
44. Byrd JC, Brown JR, O‘Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med 2014;371:213–223.
45. Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 2014;370:997–1007.
46. Coutre SE, Barrientos JC, Brown JR, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma 2015;56:2779–2786.
47. Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 2016;374:311–322.
48. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol 2016;17:768–778.
49. Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med 2018;378:1107–1120.
50. Chanan-Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol 2016;17:200–211.
51. Zelenetz AD, Barrientos JC, Brown JR, et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol 2017;18:297–311.
52. Keating MJ, Flinn I, Jain V, et al. Therapeutic role of alemtuzumab (Campath-1H) in patients who have failed fludarabine: results of a large international study. Blood 2002;99:3554–3561.
53. Stilgenbauer S, Zenz T, Winkler D, et al. Subcutaneous alemtuzumab in fludarabine-refractory chronic lymphocytic leukemia: clinical results and prognostic marker analyses from the CLL2H study of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2009;27:3994–4001.
54. Fischer K, Cramer P, Busch R, et al. Bendamustine combined with rituximab in patients with relapsed and/or refractory chronic lymphocytic leukemia: a multicenter phase II trial of the German Chronic Lymphocytic Leukemia Study Group. J Clin Oncol 2011;29:3559–3566.
55. Cuneo A, Follows G, Rigolin GM, et al. Efficacy of bendamustine and rituximab as first salvage treatment in chronic lymphocytic leukemia and indirect comparison with ibrutinib: a GIMEMA, ERIC and UK CLL FORUM study. Haematologica 2018; doi: 10.3324/haematol.2018.189837.
56. Robak T, Dmoszynska A, Solal-Celigny P, et al. Rituximab plus fludarabine and cyclophosphamide prolongs progression-free survival compared with fludarabine and cyclophosphamide alone in previously treated chronic lymphocytic leukemia. J Clin Oncol 2010;28:1756–1765.
57. Badoux XC, Keating MJ, Wang X, et al. Fludarabine, cyclophosphamide, and rituximab chemoimmunotherapy is highly effective treatment for relapsed patients with CLL. Blood 2011;117:3016–3024.
58. Awan FT, Hillmen P, Hellmann A, et al. A randomized, open-label, multicentre, phase 2/3 study to evaluate the safety and efficacy of lumiliximab in combination with fludarabine, cyclophosphamide and rituximab versus fludarabine, cyclophosphamide and rituximab alone in subjects with relapsed chronic lymphocytic leukaemia. Br J Haematol 2014;167:466–477.
59. Wierda WG, Kipps TJ, Mayer J, et al. Ofatumumab as single-agent CD20 immunotherapy in fludarabine-refractory chronic lymphocytic leukemia. J Clin Oncol 2010;28:1749–1755.
60. Durot E, Michallet AS, Lepretre S, Le QH, Leblond V, Delmer A. Platinum and high-dose cytarabine-based regimens are efficient in ultra high/high-risk chronic lymphocytic leukemia and Richter‘s syndrome: results of a French retrospective multicenter study. Eur J Haematol 2015;95:160–167.
61. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013;369:32–42.
62. Byrd JC, Furman RR, Coutre SE, et al. Three-year follow-up of treatment-naive and previously treated patients with CLL and SLL receiving single-agent ibrutinib. Blood 2015;125:2497–2506.
63. Bottcher S, Ritgen M, Fischer K, et al. Minimal residual disease quantification is an independent predictor of progression-free and overall survival in chronic lymphocytic leukemia: a multivariate analysis from the randomized GCLLSG CLL8 trial. J Clin Oncol 2012;30:980–988.
64. Logan AC, Zhang B, Narasimhan B, et al. Minimal residual disease quantification using consensus primers and high-throughput IGH sequencing predicts post-transplant relapse in chronic lymphocytic leukemia. Leukemia 2013;27:1659–1665.
65. Dreger P, Corradini P, Kimby E, et al. Indications for allogeneic stem cell transplantation in chronic lymphocytic leukemia: the EBMT transplant consensus. Leukemia 2007;21:12–17.
66. Dreger P, Schetelig J, Andersen N, et al. Managing high-risk CLL during transition to a new treatment era: stem cell transplantation or novel agents? Blood 2014;124:3841–3849.
67. Rossi D, Gaidano G. Richter syndrome: molecular insights and clinical perspectives. Hematol Oncol 2009;27:1–10.
68. Parikh SA, Kay NE, Shanafelt TD. How we treat Richter syndrome. Blood 2014;123:1647–1657.
69. Parikh SA, Shanafelt TD. Risk factors for Richter syndrome in chronic lymphocytic leukemia. Curr Hematol Malig Rep 2014;9:294–299.
70. Osterborg A, Foa R, Bezares RF, et al. Management guidelines for the use of alemtuzumab in chronic lymphocytic leukemia. Leukemia 2009;23:1980–1988.
71. Smolej L, Prochazka V, Spacek M, et al. Doporučení pro léčbu alemtuzumabem u chronické lymfocytární leukemie. Vnitr Lek 2012;58:232–236.
72. Moia R, Diop F, Favini C, Kodipad AA, Gaidano G. Potential of BCL2 as a target for chronic lymphocytic leukemia treatment. Expert Rev Hematol 2018;11:391–402.
73. Oscier D, Dearden C, Eren E, et al. Guidelines on the diagnosis, investigation and management of chronic lymphocytic leukaemia. Br J Haematol 2012;159:541–564.
74. Dhalla F, Lucas M, Schuh A, et al. Antibody deficiency secondary to chronic lymphocytic leukemia: should patients be treated with prophylactic replacement immunoglobulin? J Clin Immunol 2014;34:277–282.
75. Shanafelt T. Treatment of older patients with chronic lymphocytic leukemia: key questions and current answers. Hematology Am Soc Hematol Educ Program 2013;158–167.
76. Bokemeyer C, Aapro MS, Courdi A, et al. EORTC guidelines for the use of erythropoietic proteins in anaemic patients with cancer: 2006 update. Eur J Cancer 2007;43:258–270.
77. Aapro MS, Bohlius J, Cameron DA, et al. 2010 update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer 2011;47:8–32.
78. Tsimberidou AM, Wen S, McLaughlin P, et al. Other malignancies in chronic lymphocytic leukemia/small lymphocytic lymphoma. J Clin Oncol 2009;27:904–910.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2018 Číslo 3
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- Klinický význam menších rozestupů mezi silami přípravků s levothyroxinem v léčbě hypotyreózy
- Aktuální poznatky k léčbě gestačního diabetu a DM2 v těhotenství metforminem
- Moje zkušenosti s Magnosolvem podávaným pacientům jako profylaxe migrény a u pacientů s diagnostikovanou spazmofilní tetanií i při normomagnezémii - MUDr. Dana Pecharová, neurolog
- Nejasný stín na plicích – kazuistika
Nejčtenější v tomto čísle
- Neinfekční nemaligní lymfadenopatie – sinusová histiocytóza s masivní lymfadenopatií, nemoc Rosaiova-Dorfmanova
- Základní bioinformatické pojmy a postupy využívané pro analýzu DNA pomocí sekvenování nové generace
- Doporučení pro diagnostiku a léčbu chronické lymfocytární leukemie (CLL) – 2018
- Neinfekční a nemaligní lymfadenopatie – idiopatická (HHV-8 negativní) multicentrická forma Castlemanovy nemoci