
Nádor z blastických plazmocytoidních dendritických buněk: kazuistika a přehled literatury
Blastic plasmacytoid dendritic cell neoplasm: case report and literature review
Blastic plasmacytoid dendritic cell neoplasm represents a rare hematologic malignancy of aggressive biology and unfavourable prognosis. This disease is characterised by primary skin lesions often followed by bone marrow and extramedullary involvement. Diagnosis is based on immunohistochemistry and flow cytometric demonstration of CD4 and CD56 co-expression with variable positivity of other markers specific to dendritic cells. The median age at diagnosis ranges from 60 to 70 years, with males affected 3 times more frequently. Knowledge of its treatment is limited to retrospective analyses of small patient cohorts. Despite generally favourable treatment response to chemotherapy, the disease tends to relapse in a few months with rapid systemic dissemination. The reported data suggest that induction therapy for acute lymphoblastic leukaemia followed by allogeneic hematopoietic cell transplantation is more effective than other induction or consolidation chemotherapy strategies.
The article reports the case of a 22-year old male with newly diagnosed blastic plasmacytoid dendritic cell neoplasm with skin-limited symptoms. The patient underwent induction and one cycle of consolidation chemotherapy accordingto the GMALL protocol for acute lymphoblastic leukaemia followed by allogeneic hematopoietic cell transplantation in the first complete remission. Chemotherapy and transplantation were not associated with serious complications and the patient remains in remission with a good quality of life, 22 months after allogeneic transplantation. This case details the successful treatment strategy in a disease with unfavourable prognosis. The article also presents a review of the literature regarding diagnosis, clinical presentation and treatment strategies in patients with blastic plasmacytoid dendritic cell neoplasm. The aim of the paper is also to provide information about this rare nosological unit and promote prompt diagnosis and treatment initiation necessary for disease control.
KEY WORDS:
blastic plasmacytoid dendritic cell neoplasm – BPDCN – induction chemotherapy – GMALL – bone marrow transplantation – immunophenotyping
Autoři:
M. Čerňan 1; T. Szotkowski 1; Z. Rusiňáková 1; L. Raida 1; M. Tichý 2; M. Dušková 2
Působiště autorů:
Hemato-onkologická klinika LF UP a FN Olomouc
1; Ústav klinické a molekulární patologie LF UP a FN Olomouc
2
Vyšlo v časopise:
Transfuze Hematol. dnes,23, 2017, No. 3, p. 134-144.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Souhrn
Nádory z blastických plazmocytoidních dendritických buněk představují raritní hematologickou malignitu s agresivním průběhem a nepříznivou prognózou. Pro onemocnění jsou charakteristické primární kožní léze, často se současným postižením kostní dřeně a extramedulárních orgánů. Diagnostika je založena na imunohistochemickém průkazu a potvrzení ko-exprese znaků CD4 a CD56 s variabilním vyjádřením dalších markerů specifických pro dendritické buňky pomocí průtokové cytometrie. Medián věku v době diagnózy je 60–70 let, onemocnění se 3krát častěji vyskytuje u mužů. Poznatky o léčbě jsou omezeny na retrospektivní analýzy malých souborů nemocných. Navzdory obecně dobré iniciální léčebné odpovědi na chemoterapii dochází v odstupu řádu měsíců k časnému relapsu a následné rychlé systémové diseminaci. Publikovaná data ukazují na vyšší účinnost indukční léčby pro akutní lymfoblastickou leukemii následovanou alogenní transplantací krvetvorných buněk ve srovnání s jinými režimy indukční, respektive konsolidační chemoterapie.
Předkládaný článek přináší kazuistiku 22letého nemocného, u něhož byla stanovena diagnóza nádoru z plazmocytoidních dendritických buněk s izolovaným kožním postižením. Pacient podstoupil indukční a jeden cyklus konsolidační chemoterapie podle protokolu GMALL pro akutní lymfoblastickou leukemii, následovanou alogenní transplantací krvetvorných buněk od nepříbuzenského dárce v 1. kompletní remisi onemocnění. Podání chemoterapie i provedení transplantace proběhlo bez závažných komplikací a nemocný zůstává v remisi onemocnění s dobrou kvalitou života v době vzniku této práce, 22 měsíců od alogenní transplantace. Kazuistika dokumentuje úspěšný léčebný přístup k prognosticky nepříznivému onemocnění. Článek je doplněný o literární přehled diagnostiky, klinického obrazu a léčebných strategií u pacientů s nádory z blastických plazmocytoidních dendritických buněk. Cílem sdělení je také zvýšit povědomí o této vzácné nozologické jednotce, a přispět tak k rychlejší diagnostice a včasnému zahájení intenzivní terapie nutné ke kontrole průběhu nemoci.
KLÍČOVÁ SLOVA:
nádor z blastických plazmocytoidních dendritických buněk – BPDCN – indukční chemoterapie – GMALL – transplantace kostní dřeně – imunofenotypizace
ÚVOD
Nádory z blastických plazmocytoidních dendritických buněk (BPDCN – Blastic Plasmacytoid Dendritic Cell Neoplasm) představují vzácné maligní onemocnění krvetvorby vycházející z prekurzorů plazmocytoidní dendritické buňky, s častým primárním postižením kůže a tendencí k rychlé systémové diseminaci [1]. Pojmenování a diagnostická kritéria této vzácné klinické jednotky prodělala v průběhu let řadu změn. Poprvé bylo onemocnění popsáno v literatuře v roce 1994 jako “kožní lymfom s izolovanou pozitivitou CD4 a CD56” [2]. Pro agranulární morfologii blastů se specifickým imunofenotypem (CD4+, CD56+, CD15+ a CD3-) označili Brody et al. toto onemocnění o rok později jako “agranulární CD4+ NK (natural killer) leukemii” [3]. V roce 2002 navrhli Petrela et al. na základě charakteristického imunofenotypu a predilekčního kožního postižení název “agranulární CD4+ CD56+ hematodermické neoplazma” [4]. Čtvrté vydání WHO (World Health Organization) klasifikace myeloidních neoplazií a akutních leukemií z roku 2008 zavedlo pro malignitu vycházející z plazmocytoidních dendritických buněk (typu DC 2) s charakteristickým klinickým průběhem označení “nádor z blastických plazmocytoidních dendritických buněk”. Nová nozologická jednotka byla současně v klasifikaci zařazena do skupiny akutních myeloidních leukemií (AML) a přidružených neoplazií [5]. Revize WHO klasifikace z roku 2016 nepřinesla žádné zásadní změny v pojmenování ani diagnostických kritériích BPDCN [6].
KAZUISTIKA
Popis případu
Prezentujeme případ 22letého nemocného, který byl v únoru 2015 přijat na naši kliniku k terapii pro nově stanovenou diagnózu nádoru z blastických plazmocytoidních dendritických buněk. Pacient byl vstupně vyšetřen ve spádové nemocnici pro výsev exantému v oblasti obličeje a zad. Kožní eflorescence měly charakter vícečetných lividních, nebolestivých, papulózních útvarů s průměrem do 2 cm. V iniciální diferenciální diagnostice byl zvažován aterom a nemocný podstoupil probatorní excizi z ložiska v lumbální oblasti. Ze vzorku byl odečten (konfirmační druhé čtení) nádor typu BPDCN. Pacient byl s nálezem následně odeslán k došetření a zahájení terapie na naše pracoviště.
Diagnostika
Při vstupním vyšetření, přibližně 2 měsíce od první manifestace kožních lézí, byla nalezena makulózní, pigmentovaná, nebolestivá kožní ložiska v oblasti trupu a obličeje s průměrem přibližně 2 cm, největší morfa v oblasti levé lopatky s průměrem asi 3 cm. Nemocný negoval přítomnost systémových příznaků onemocnění – hmotnostní úbytek, noční pocení nebo horečky. V klinickém obraze dominovalo kožní postižení bez nálezu hepatomegalie, splenomegalie či lymfadenomegalie. PET/CT vyšetření prokázalo zvýšenou akumulaci glukózy v zesílené kožní vrstvě na zádech (v místě první probatorní biopsie) a nad levou lopatkou, v místě největší kožní morfy. Vyšetření krevního obrazu (hemoglobin 164 g/l, trombocyty 332 x 109/l, leukocyty 8,05 x 109/l – s normálním diferenciálním rozpočtem, bez nálezu blastů) ani vyšetření biochemie či koagulace neprokázaly odchylky od normálních laboratorních hodnot. Vzhledem ke vzácnosti diagnózy byla provedena nová biopsie z největší kožní morfy nad levou lopatkou s nálezem infiltrátu z blastických elementů s imunofenotypem CD56, CD4 a CD43, TdT (terminální deoxynukleotidyltransferáza) a LCA (30 %) současně s negativitou markerů CD1, CD7 a CD68. Konfirmační imunohistochemické vyšetření z druhé biopsie potvrdilo vstupní diagnózu BPDCN. Cytogenetická analýza nádorových buněk z kožní léze prokázala komplexní změny karyotypu zahrnující – monosomii 9, monosomii 13, t(1;9;1)(q21;q21;p35), t(6;7;7)(q21;q21;p13), ins(12;5)(p?13;q?) a t(6;11)(q23;q24). Trepanobiopsie z lopaty kosti kyčelní ani imunofenotypizační vyšetření kostní dřeně a mozkomíšního moku neprokázala infiltraci nádorovými buňkami s imunofenotypem BPDCN. Obrázek 1 ukazuje imunohistochemické barvení řezu z kožní biopsie na markery specifické pro BPDCN.

A: Infiltrát blastických elementů v dermis (HE, 200x).
B: Membránová exprese CD56 (anti-CD56, 200x).
C: Membránová exprese CD4 (anti-CD4, 200x).
D: Exprese terminální deoxynukleotidyltransferázy v jádrech blastických buněk (anti-TdT, 200x).
(Zdroj: Ústav klinické a molekulární patologie LF UP a FN Olomouc)
Indukční a konsolidační chemoterapie
Vzhledem k velmi dobrému biologickému stavu (bez jakýchkoliv komorbidit) a nízkému věku byl pacient na základě publikovaných dat indikován k intenzivní chemoterapii následovanou alogenní transplantací krvetvorných buněk (TKB). Pro podezření na klinickou progresi onemocnění – indurace kožních ložisek, byla od března 2015 zahájena předfáze kortikosteroidy (dexametazon 2krát 8 mg denně po dobu 5 dnů). Obrázky 2 a 3 ukazují kožní léze na hrudníku po zahájení předfáze kortikosteroidy. Následně byla za hospitalizace od března 2015 podána 1. fáze indukční chemoterapie pro akutní lymfoblastickou leukemii (ALL) podle protokolu GMALL ALL modifikace CELL 2012 (dexametazon, cyklofosfamid, vinkristin, daunorubicin, pegylovaná L-asparagináza). Systémová chemoterapie byla doplněna profylaktickou intratekální chemoterapií (cytarabin 40 mg, metotrexát 15 mg a dexametazon 4 mg) podle protokolu. Nemocný snášel podání 1. cyklu léčby bez komplikací, po podání chemoterapie došlo ke kompletní regresi kožních ložisek. Od dubna 2015 byla pak podána 2. fáze indukční chemoterapie (cyklofosfamid, cytarabin, 6-merkaptopurin), doplněna o 3 profylaktické intratekální aplikace chemoterapie identického složení jako při 1. cyklu léčby. Podání 2. fáze indukce bylo komplikováno polékovou hepatopatií s pozvolnou úpravou na symptomatické terapii. Následně byla od června 2015 podána 1. konsolidace (dexametazon, vindesin, vysoce dávkovaný metotrexát, etoposid, vysoce dávkovaný cytarabin) s profylaktickou intratekální aplikací. Třetí cyklus léčby byl komplikovaný přechodnou stomatitidou s odynofagií. Nemocný snášel 3 cykly léčby podle uvedeného protokolu bez závažných komplikací.

(Zdroj: Hemato-onkologická klinika LF UP a FN Olomouc)
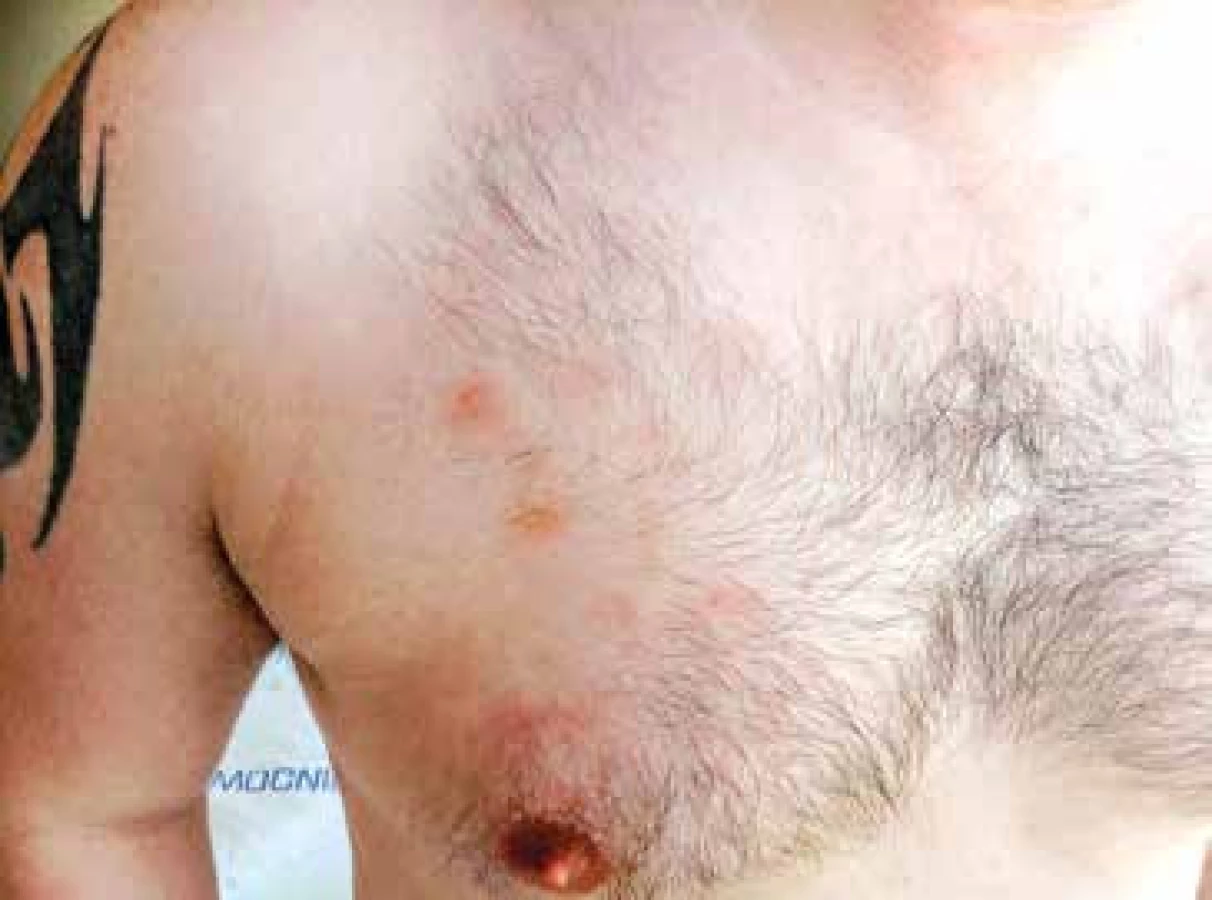
(Zdroj: Hemato-onkologická klinika LF UP a FN Olomouc)
Transplantace krvetvorných buněk
HLA-typizační vyšetření sestry nemocného neprokázalo shodu s pacientem, proto bylo zahájeno vyhledávání v registrech dárců kostní dřeně. Po dokončení standardních předtransplantačních vyšetření byl nemocný v červenci 2015 přijat k provedení alogenní TKB od nepříbuzenského dárce, 23letého muže, HLA inkompatibilního (1 neshoda na lokusu A), AB0 inkompatibilního (příjemce B+, dárce 0+), CMV negativního. Popřípravném režimu s redukovanou intenzitou (RIC) Flu + Mel + TG (fludarabin 150 mg/m2, melfalan 140 mg/m2, thymoglobulin 4,0 mg/kg), byl 23. 7. 2015 proveden převod štěpu s obsahem 4,29 x 106/kg CD34+ a 2,65 x 108/kg jaderných buněk. Komplikací v potransplantačním období byl rozvoj klostridiové enteritidy (pozitivní klostridiový toxin i antigen) s promptním ústupem potíží na terapii metronidazolem per os. Další průběh hospitalizace byl již bez potíží. Nemocný byl propuštěn v den +25 po transplantaci ve stabilizovaném stavu do ambulantní péče. Prevence GVHD (graft-versus-host disease) byla zajištěna kombinací cyklosporinu A (CyA) a mykofenolát mofetilu (MMF). Aplikace CyA byla zahájena v den -1 před transplantací v dávce 6 mg/kg denně a následně korigována, s cílem udržet v časném potransplantačním období hladinu 200–300 ng/ml. Při absenci jakýchkoliv známek GVHD byla redukce CyA zahájena v den +56 s cílem pozvolného vysazení do dne +110 až 130 po TKB při remisi základního onemocnění a 100% dárcovském buněčném chimérismu. Podávání MMF bylo zahájeno v den +1 v dávce 30 mg/kg/den, s redukcí a postupným vysazením mezi dny +29 a +56.
Ambulantní sledování
Ode dne +25 byla opakovaně detekována EBV-DNA v plazmě a aspirátu kostní dřeně, ve které byl současně zaznamenán postupný nárůst až k titru 231 x 103 kopií/ ml v den +77 spolu s mírným poklesem dárcovské chiméry v jaderných buňkách (ze 100 % na 97 %). Pro vysoké riziko rozvoje potransplantační EBV asociované lymfoproliferace byl nemocnému v den +89 preemptivně podán rituximab v celkové dávce 700 mg. Současně bylo urychleno vysazování CyA, s poslední aplikací v den +98 po TKB. Kontrolní vyšetření po uvedených intervencích již neprokázalo EBV-DNA v plazmě ani aspirátu kostní dřeně s opětovným a zatím přetrvávajícím dosažením 100% dárcovského buněčného chimérismu. Pro hypogamaglobulinemii byly nemocnému opakovaně ambulantně aplikovány imunoglobuliny. Pacient zůstává i nadále dispenzarizován v ambulanci pro transplantované nemocné.
Shrnutí
Kazuistika popisuje případ mladého nemocného s nádorem z blastických plazmocytoidních dendritických buněk. Diagnóza byla u pacienta stanovena v horizontu 2 měsíců od prvních příznaků onemocnění. Vstupní staging prokázal jen limitované kožní postižení, bez infiltrace kostní dřeně a extramedulárních orgánů. Nemocný podstoupil indukční léčbu a jeden cyklus konsolidační léčby podle protokolu pro ALL včetně intratekální profylaktické chemoterapie. Vzhledem k obecně nepříznivé biologii onemocnění s tendencí k časnému relapsu, rychlé systémové diseminaci a s přihlédnutím k nepříznivému cytogenetickému nálezu byla u pacienta provedena alogenní TKB v první kompletní remisi onemocnění, v souladu s daty publikovanými v literatuře, a to i přes dostupnost pouze nepříbuzného HLA inkompatibilního dárce. V době přípravy rukopisu (květen 2017) byl pacient 22 měsíců po alogenní TKB v trvající remisi onemocnění, bez projevů chronické reakce štěpu proti hostiteli (GVHD) a s dobrou kvalitou života, včetně návratu k původní profesi.
EPIDEMIOLOGIE
Skutečná incidence onemocnění není známa, předpokládá se, že BPDCN představují asi 0,44 % všech hematologických malignit, méně než 1 % případů vstupně se manifestujících jako akutní leukemie a 0,7 % kožních lymfomů [7–9]. Onemocnění se častěji vyskytuje u starších nemocných s mediánem věku 60–70 let. Přibližně jen 5 % případů bylo popsáno u dětí mladších 10 let. Literatura uvádí až 2–3krát vyšší incidenci onemocnění u mužského pohlaví [1, 10–12]. Zatím nebyly identifikovány žádné environmentální nebo hereditární faktory predisponující ke vzniku tohoto onemocnění. Přibližně u 10–20 % nemocných je známa anamnéza předchozí malignity [1, 12]. Případy sekundární BPDCN byly popsány v návaznosti na chemoterapii (± radioterapii) pro některé solitární tumory (karcinom prostaty, prsu) a hematologické malignity – nehodgkinské lymfomy, akutní lymfoblastickou leukemii či chronickou myelomonocytární leukemii [12, 13]. Pagano et al. popsali 4 případy sekundární BPDCN, kterým předcházela diagnóza myelodysplastického syndromu (MDS), přičemž všichni nemocní dostávali jen podpůrnou péči pro základní diagnózu. Vztah mezi BPDCN a MDS není zatím jasně vysvětlen, asociace obou onemocnění může vycházet z původu neoplastického klonu v myeloidní linii [12].
LABORATORNÍ NÁLEZY A DIAGNOSTICKÁ KRITÉRIA
Imunofenotypizace a imunohistochemie
Navzdory pokrokům ve vyšetřovacích metodách i novelizacím kritérií zůstává diagnostika BPDCN i nadále problematická. Stanovení diagnózy je založeno na průkazu charakteristického imunofenotypu nádorových buněk – imunohistochemicky nebo průtokovou cytometrií, v závislosti na dostupnosti biologického materiálu. Charakteristická pro diagnózu je ko-exprese znaků CD4 a CD56 s variabilním vyjádřením dalších markerů specifických pro dendritické buňky – CD123, CD43, BDCA-2/CD303, TCL1, při současné negativitě liniově charakteristických znaků pro B a T lymfocytární, myeloidní, monocytární linii a NK buňky. Znaky CD5, CD7, CD33 a TdT (terminální deoxynukleotidyltransferáza) mohou být variabilně vyjádřeny [5, 10]. V literatuře byly popsány i případy BPDCN s negativitou znaků CD4 nebo CD56 a současně popsány i jiné hematologické malignity s pozitivitou obou uvedených markerů (extranodální CD56+ NK/T lymfom, kožní T lymfom, nediferencovaná AML, obtížně klasifikovatelná akutní leukemie – acute leukaemia of ambiguous lineage), proto je pro stanovení diagnózy vyžadována přítomnost i dalších znaků charakteristických pro plazmocytoidní dendritické buňky [1, 10]. Zatím nebyl stanoven minimální rozsah imunofenotypových vyšetření potřebných ke stanovení diagnózy. Analýza souborů nemocných publikovaných v literatuře však ukázala na vysokou specificitu pozitivity alespoň 4 znaků ze skupiny CD4, CD56, CD123, TCL1 a CD303 [10, 14].
Morfologie
Tumor z blastických plazmocytoidních dendritických buněk je tvořen monomorfními buňkami připomínajícími lymfoblasty nebo myeloblasty. Cytoplazma bývá redukovaná, šedomodré barvy, postrádající azurofilní granula v barvení podle May-Grünwald-Giemsy. V cytoplazmě mohou být přítomny vakuoly a pseudopodiální výběžky. Jadérko je nepravidelného tvaru s výrazným chromatinem. Hodnota indexu Ki-67 bývá různá, v rozmezí 20–80 %. Kožní léze vykazují masivní blastickou infiltraci dermis až do podkožního tuku. Epidermis a kožní adnexa nebývají postižena [10, 13].
Cytogenetika a molekulární biologie
Zatím nebyla nalezena žádná specifická cytogenetická změna patognomická pro BPDCN. Častým nálezem jsou komplexní změny karyotypu [10–12]. Ve skupině 21 nemocných Leroux et al. identifikovali 6 chromozomů s častým výskytem aberací – 5q (72 %), 12p (64 %), 13q (64 %), 6q (50 %), 15q (43 %) a monosomie 9 (28 %) [15]. Aberace na uvedených chromozomech nalezli u 8 z 11 nemocných s dostupným cytogenetickým vyšetřením i Tsagarakis et al. [13]. Bialelická ztráta lokusu 9p21.3 byla u nemocných s BPDCN asociována se signifikantně kratším celkovým přežitím ve srovnání s hemizygotní ztrátou (11 vs. 26 měsíců, p = 0,0349) [16]. Pagano et al. ve skupině 28 nemocných s dostupným cytogenetickým vyšetřením zaznamenali normální karyotyp u 13 (46 %) a komplexní změny karyotypu u 10 (36 %) nemocných. Tři nemocní s normálním cytogenetickým nálezem měli FLT3-ITD mutaci [12]. Metodou NGS (next-generation sequencing) byl ve skupině 7 nemocných zjišťován výskyt 219 mutací rekurentně se vyskytujících u hematologických malignit – výsledky potvrdily výskyt TET2, TP53, ASXL1 a ZRSR2 mutací u BPDCN [17]. U pacientů s nádorem z plastických plazmocytoidních dendritických buněk může být nalezeno široké spektrum genetických abnormalit, některé by v budoucnu mohly sloužit k prognostické stratifikaci i jako terapeutické cíle pro nové léky.
KLINICKÝ OBRAZ
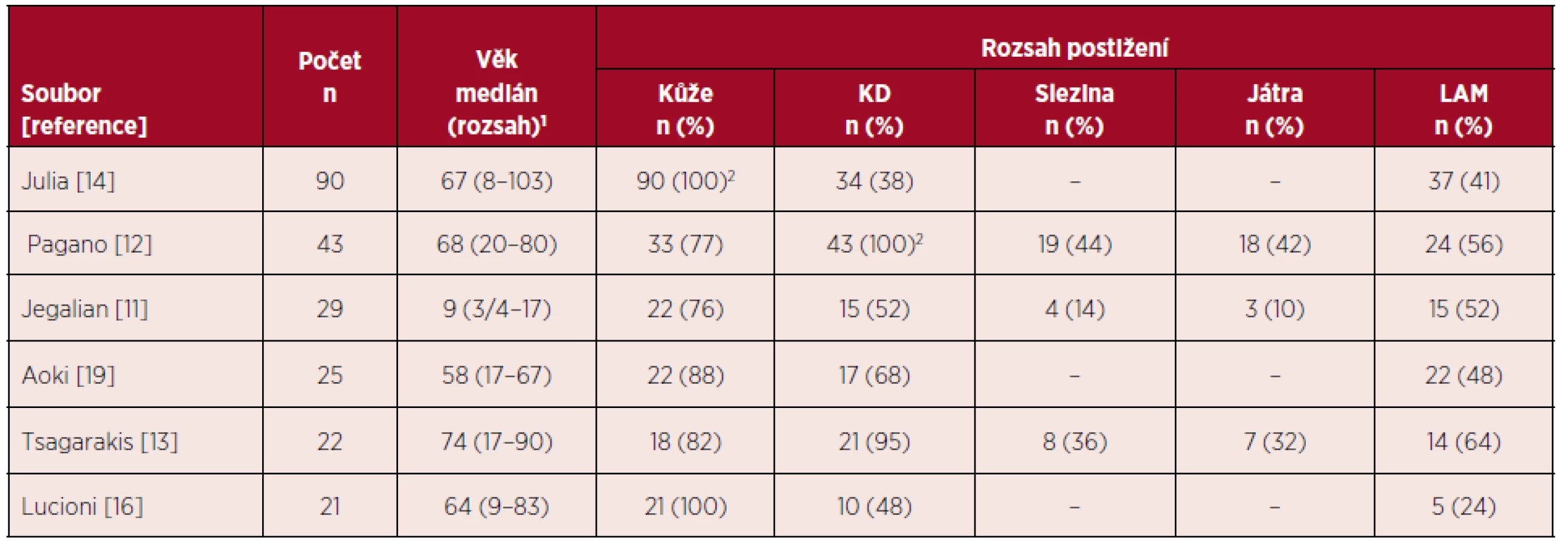
BPDCN je vzácná hematologická malignita s charakteristickým klinickým obrazem a průběhem. Navzdory často iniciálně indolentní manifestaci má onemocnění sklon k rychlé systémové diseminaci s infiltrací kostní dřeně a extramedulárních orgánů [5, 18]. Mezi první příznaky onemocnění patří kožní postižení, které je při stanovení diagnózy přítomno u více než 85 % nemocných [11–14, 19]. Rozsah a charakter kožních lézí může být značně variabilní. Ve skupině 90 nemocných s BPDCN měly kožní léze charakter nodulací u 63 (77 %) nemocných, u 11 (12 %) vzhled podlitin a smíšený obraz (makuly a noduly) u 13 pacientů (14 %). Pět nemocných mělo současně přítomno slizniční postižení [14]. V době stanovení diagnózy je kožní postižení manifestní u většiny nemocných, až 80 % pacientů má však současně postižení dalších orgánů – kostní dřeně (> 50 %), lymfatických uzlin (> 50 %), sleziny a jater (11–20 %) [18]. Jiné orgány bývají postiženy výrazně méně častěji – nosohltan, tonzily, CNS nebo svaly. Infiltrace CNS bývá popisována u 4–9 % nemocných při stanovení diagnózy a u 17–33 % nemocných při progresi nebo relapsu onemocnění [1, 10, 12, 13]. V literatuře byly referovány i případy vstupně bez kožního postižení, manifestující se pod obrazem akutní leukemie s imunofenotypem blastických plazmocytoidních dendritických buněk. Postižení kostní dřeně maligními buňkami vede k útlaku fyziologické krvetvorby s chyběním zralých elementů v periferní krvi – častým nálezem je pak anémie, trombocytopenie a neutropenie, s klinickým obrazem podobným AML či ALL [12]. Hyperleukocytóza nepatří k častým nálezům, přítomnost blastů v periferní krvi je však mnohem častější [10]. Na rozdíl od lymfoidních malignit jsou B-symptomy jako horečka, noční pocení a hubnutí u BPDCN vzácné [18]. Tabulka 1 přináší přehled klinických nálezů při stanovení diagnózy ve vybraných souborech nemocných s BPDCN.
PROGNOSTICKÉ FAKTORY
Prognóza onemocnění navzdory pokrokům v diagnostice a terapii zůstává nepříznivá, s mediánem celkového přežití 9–15 měsíců [11–13, 18]. Je obtížné stanovit prognostické faktory, jelikož onemocnění má velmi nízkou incidenci, značně heterogenní rozsah postižení (kožní léze ± infiltrace kostní dřeně a extramedulárních orgánů) a široké spektrum používaných léčebných přístupů. Vyšší věk a nedosažení kompletní remise onemocnění jsou považovány za nepříznivé prognostické faktory. Ve skupině 43 nemocných s BPDCN byl medián celkového přežití signifikantně delší u nemocných mladších 65 let (12,6 vs. 7,1 měsíce, p = 0,04) a při dosažení kompletní remise onemocnění (14 vs. 4,5 měsíce, p < 0,0001) [12]. Lepší výsledky léčby a delší celkové přežití ve srovnání s dospělými pacienty byly zaznamenány v souboru dětských nemocných [11]. Izolovaná kožní manifestace onemocnění byla v některých studiích u dospělých vyhodnocena jako prognosticky příznivá. Nemocní s izolovaným kožním postižením měli statisticky nevýznamně delší medián celkového přežití ve srovnání s nemocnými se systémovým postižením (17 vs. 12 měsíců, p > 0,05) [16, 18]. Bialelická ztráta lokusu 9p21.3 byla asociována se signifikantně kratším celkovým přežitím (11 vs. 26 měsíců, p = 0,0349) [16]. Zásadní pro prognózu je i výběr léčebné strategie. Publikovaná data ukazují na lepší výsledky léčby při použití intenzivních léčebných protokolů pro ALL, následovaných alogenní transplantací krvetvorných buněk ve srovnání s non-intenzivní, případně jen lokální terapií [12, 13, 18]. Lokální ozáření a chirurgická excize ložisek u nemocných s limitovaným kožním postižením vedla jen ke krátkodobé remisi onemocnění, s mediánem 5,5 měsíce do relapsu [20].
TERAPIE
Poznatky o léčbě tumorů z blastických plazmocytoidních dendritických buněk se v literatuře vzhledem k nízké incidenci onemocnění objevují jen velmi zřídka. Dosud chybí doporučené postupy i rozsáhlejší prospektivní studie. Retrospektivní analýzy publikovaných souborů nemocných ukazují na vyšší počet dosažených kompletních remisí (KR) a delší celkové přežití při použití indukční chemoterapie pro akutní lymfoblastické leukemie, následované alogenní transplantací krvetvorných buněk ve srovnání s jinými režimy indukční, respektive konsolidační chemoterapie [12, 13, 16, 18].
Indukční léčba
Ve skupině 43 nemocných s BPDCN manifestujících se v době diagnózy jako akutní leukemie, byla KR dosažena signifikantně častěji po indukční chemoterapii pro ALL/lymfomy ve srovnání s indukcí pro AML (67 % vs. 27 %, p = 0,02). Medián celkového přežití byl v souboru 8,7 měsíce, přičemž nemocní podstupující chemoterapii používanou u agresivních lymfoproliferací měli medián celkového přežití významně delší než po indukci pro AML (12,3 vs. 7,1 měsíce, p = 0,02). Alogenní TKB měla signifikantní vliv na delší celkové přežití nemocných ve srovnání s léčbou omezenou na chemoterapii (22,7 vs. 7,1 měsíce, p = 0,03) [12]. Významně delší celkové přežití u transplantovaných nemocných (31,3 vs. 12 měsíců, p > 0,05) popsali i Dalle et al. [20]. Výsledky terapie u 86 nemocných s BPDCN léčených ve francouzských centrech referovali Poret et al. Indukční léčbu pro ALL podstoupilo 17, pro AML 19 pacientů a shodně 16 nemocných dostalo léčbu pro NK/T-lymfoproliferace (vysokodávkovaný metotrexát + L-asparagináza ± dexametazon) a chemoterapii podle protokolu CHOP. Kompletní remise byla dosažena u 70,6 % a 78,9 % nemocných po indukci pro akutní leukemie a u 62,5 %, respektive 31,3 % nemocných dostávajících další režimy. Medián celkového přežití u 34 nemocných, kteří podstoupili autologní nebo alogenní TKB byl signifikantně delší než u 52 netransplantovaných pacientů v souboru (49 vs. 8 měsíců, p < 0,0001) [21]. Tsagarakis et al. ve skupině 22 nemocných popsali dosažení KR u všech 9 nemocných s BPDCN, kteří dostali indukci pro ALL, u 3/6 (50 %) po AML a 3/4 (75 %) nemocných dostávajících léčbu podle protokolů pro lymfomy. Navzdory iniciálně dobré odpovědi na léčbu – 15/19 (79 %) nemocných dosáhlo KR a 5 následně podstoupilo alogenní TKB v první remisi onemocnění – 2leté přežití v souboru bylo jen 43 % [13]. V souboru 29 nemocných s BPDCN mladších 18 let popsali Jegalian et al. dosažení léčebné odpovědi u všech 14 pacientů dostávajících indukci pro ALL (12 KR a 2 parciální remise). Naopak 3 nemocní, kteří dostali indukci pro AML zemřeli (1 nemocný na progresi onemocnění a další 2 na komplikace léčby). Základem terapie BPBCN u dětí by měla být chemoterapie pro ALL s profylaxí CNS postižení a indikací TKB u nemocných s relapsem nebo jen parciální odpovědí na 1. linii léčby [11]. HyperCVAD (vysokofrakcionovaný cyklofosfamid, vinkristin, adriamycin a dexametazon) alternující s kombinací metotrexát a cytarabin vedli k dosažení KR u 11/12 (90 %) nemocných s mediánem trvání 21 (4–39) a mediánem celkového přežití 29 (1–44) měsíců [22]. V souborech nemocných s BPDCN publikovaných v literatuře jsou relativně častěji popisovány případy postižení CNS, jehož terapie by měla být vedena podle příslušného ALL protokolu [12].
Transplantace krvetvorných buněk
Podobně jako u jiných hematologických malignit s agresivním chováním a tendencí k systémové diseminaci má alogenní transplantace v 1. kompletní remisi podobný význam i u BPDCN [18]. Signifikantně delší celkové přežití u transplantovaných nemocných bylo potvrzeno ve více publikovaných souborech nemocných [12, 13, 20]. Alogenní TKB po RIC by měla být zvažována i u starších pacientů. Indikace autologní TKB je rezervována jen pro nemocné s chemosenzitivní chorobou (v rámci časného managementu onemocnění) nebo u starších nemocných v dobrém celkovém stavu nebo při nedostupnosti dárce kostní dřeně. Je nepravděpodobné, že by autologní TKB mohla vést k dlouhodobé remisi u nemocných s floridním nebo relabujícím/refrakterním onemocněním [18, 19]. Efektivita autologní TKB byla potvrzena ve skupině 11 nemocných s mediánem věku 57 let s dosažením celkového 4letého přežití 82 % [19]. Vzhledem k častému postižení CNS při stanovení diagnózy nebo relapsu by měly být při autologní TKB použity přípravné režimy s thiotepou (s dobrou přestupností přes hematoencefalickou bariéru) používané u agresivních lymfomů [1]. Tabulka 2 přehledně zobrazuje výsledky léčby ve vybraných souborech nemocných s BPDCN.

Neintenzivní léčba
U nemocných s izolovaným kožním postižením může lokální terapie (chirurgické odstranění, fokální radioterapie) doplněná o systémové podání kortikosteroidů nebo nízko dávkovanou chemoterapii vést ke krátkodobé remisi onemocnění v trvání 6–9 měsíců. Non-intenzivní přístupy tak mohou představovat paliativní modalitu u nemocných neschopných podstoupit intenzivní chemoterapii [20].
Klinické studie a nové léky
První prospektivní klinická studie u nemocných s BPDCN prokázala efektivitu rekombinantního proteinu s označením SL-401, skládajícího se z difterického toxinu s cytotoxickým efektem navázaného na IL-3 (interleukin 3). Mechanismus účinku SL-401 je daný vysokou mírou exprese receptorů pro IL-3 na nádorových buňkách. Jedenáct nemocných s mediánem věku 70 let dostávalo studiovou dávku 12,5 μg/kg SL-401 po dobu 5 dní. Kompletní remisi dosáhlo 5 pacientů (45 %) a parciální remisi 2 nemocní (18 %) [23, 24]. Jsou potřebné další studie na větších souborech pacientů a v kombinaci s chemoterapií k potvrzení efektivity a stanovení pozice SL-401 v managementu terapie BPDCN. Vzhledem k popsané přítomnosti FLT3-ITD mutace, mohou FLT3 inhibitory představovat další terapeutickou modalitu podobně jako u FLT3-ITD pozitivní AML [12,25]. Obrázek 4 ukazuje algoritmus terapie BPDCN.

CHT – chemoterapie, KS – kortikosteroidy, RT – radioterapie, ALL – akutní lymfoblastická leukemie,
AML – akutní myeloidní leukemie, TKB – transplantace krvetvorných buněk
(volně upraveno podle Pagano et al., 2016)
DISKUSE
Nádory z blastických plazmocytoidních dendritických buněk představují závažné onemocnění s nepříznivou prognózou. Plazmocytoidní dendritické buňky (typu DC2) mají spolu s myeloidními dendritickými buňkami (typu DC1) původ ve společném progenitoru v kostní dřeni. Na rozdíl od myeloidních dendritických buněk, které patří mezi fagocyty a antigen-prezentující buňky imunitního systému se plazmocytoidní dendritické buňky vyznačují především produkcí zánětlivých cytokinů (interferony typu 1), aktivací NK buněk a mechanismů vrozené imunity. Historická označení této vzácné klinické jednotky vycházela ze současných poznatků na poli morfologie, imunohistochemie a imunologie. Aktuálně používaná nomenklatura poukazuje na původ nádoru ve specifické buněčné populaci typu DC2 s charakteristickým imunohistochemickým profilem, odlišným od jiných vývojových linií.
Incidence onemocnění je vyšší u mužského pohlaví a starších nemocných s mediánem věku přibližně 65 let [12, 14, 18, 19]. Soubory pacientů publikované v literatuře popisují často jen jednotky, výjimečně desítky případů onemocnění. V naší literatuře byly zatím referovány 2 kazuistiky – 64letý pacient s izolovaným kožním postižením a 18letá nemocná s akutní leukemií s imunofenotypem BPDCN [26, 27]. Námi prezentovaná kazuistika je třetím publikovaným případem onemocnění v České republice.
Klinický obraz BPDCN vykazuje charakteristické chování, iniciálně s predominantním kožním postižením a tendencí k systémové diseminaci při progresi či relapsu onemocnění [1]. Diagnostika může být zpočátku pro indolentní průběh a jen limitované kožní či systémové projevy značně obtížná. Prvním příznakem nutícím pacienty k vyhledání lékařské péče bývá kožní postižení. Ve skupině 90 nemocných s kožními lézemi byly vstupně zaznamenány limitované příznaky (1–2 nodulární léze) u 42 (47 %) pacientů. Medián času do stanovení diagnózy od prvních příznaků byl v souboru 6,2 měsíce [14]. V případě našeho nemocného byla první manifestace onemocnění ve formě izolovaného kožního postižení. Vstupně zvažovaná diagnóza ateromů se nepotvrdila, imunohistochemické vyšetření prokázalo diagnózu BPDCN. Diagnóza byla stanovena do 2 měsíců od prvních příznaků nemoci. Jednalo se o vzácné onemocnění s nepříznivou prognózou a náročnou léčbou, proto bylo vzhledem k iniciálně indolentnímu průběhu rozhodnuto o provedení rebiopsie. Při podezření na BPDCN by nemocný měl být odeslán na specializované pracoviště s dostupností imunohistochemických metod a metod průtokové cytometrie, které jsou pro verifikaci diagnózy stěžejní. Minimální rozsah vyšetření pro stanovení diagnózy BPDCN je pozitivita znaků CD4, CD56 a CD 123 při negativitě liniově specifických znaků. Vysoce specifické markery pro plazmocytoidní dendritické buňky – BDCA2/CD303, TCL1 a CD2AP podporují diagnózu BPDCN [12,20]. Dostatečně široký rozsah imunofenotypizačního vyšetření může přispět k včasné identifikaci nemocných s BPDCN a současně k odlišení onemocnění s částečně shodným imunofenotypovým profilem – extranodální CD56+ NK/T lymfom, kožní T-buněčný lymfom nebo obtížně klasifikovatelná akutní leukémie [1]. Při nesplnění imunofenotypových diagnostických kritérií pro BPDCN podle WHO klasifikace má být onemocnění s obrazem akutní leukemie klasifikováno jako akutní leukemie obtížně klasifikovatelná (acute leukaemia of ambiguous lineage) [1, 5].
Výsledky terapie v souborech nemocných s BPDCN publikovaných v literatuře poukazují na vyšší počet dosažených kompletních remisí použitím indukční chemoterapie pro ALL a delší celkové přežití u alogenně transplantovaných nemocných. Použití protokolů pro ALL s širším spektrem cytostatik v prvním cyklu chemoterapie se zdá být logičtější než protokoly pro léčbu AML. Avšak vzhledem k původu onemocnění v myeloidním prekurzoru Pagano et al. doporučují konsolidační chemoterapii se středně/vysokodávkovaným cytarabinem, analogicky léčbě AML [1]. Poret et al. navrhují jako základ pro léčbu BPDCN protokol kombinující metotrexát, idarubicin, L-asparaginázu a dexametazon, tj. léky, které prokázaly svoji efektivitu v retrospektivních analýzách. Alogenní TKB je následně indikována v 1. kompletní remisi onemocnění u jinak zdravých pacientů. V případě neschopnosti podstoupit alogenní TKB (věk, komorbidity, nedostupnost dárce) by měly být podány 3 cykly chemoterapie, s rotací uvedených léků a následně provedená autologní TKB [21]. Vzhledem k tomu, že alogenně transplantovaní nemocní mají mnohem lepší dlouhodobé výsledky, je potřeba zvážit transplantaci i u starších pacientů po přípravných režimech s redukovanou intenzitou [12]. Radioterapie či chirurgická excize u lokalizovaného kožního postižení vede jen ke krátkodobé remisi. Onemocnění je již vstupně považováno za generalizovanou malignitu vyžadující systémové podání chemoterapie [20]. Vzhledem k časté infiltraci CNS, pohybující se v souborech nemocných od 4 až do 33 % při stanovení diagnózy, respektive relapsu/progresi onemocnění, by intratekální profylaxe měla být součástí léčebného protokolu [1, 12, 13]. Pagano et al. zaznamenali 3 izolované CNS relapsy ve skupině 17 nemocných, kteří dosáhli kompletní remise po indukční léčbě. Ani jeden z nemocných nedostal CNS profylaxi [12].
Lepší výsledky léčby u mladších nemocných a dětí mohou vycházet z možnosti podání agresivnější chemoterapie a schopnosti podstoupit alogenní TKB. Je však nutné zvážit i možnost odlišné biologie základního onemocnění, s rozdílným chováním v dětském a dospělém věku, podobně jako u ALL. Alogenní TKB by u dětí měla být indikována jen v případě nedostatečné odpovědi na 1. linii léčby nebo při relapsu onemocnění [11]. Navzdory pokrokům v léčbě a diagnostice BPDCN zůstavá prognóza většiny nemocných zatím nepříznivá, s mediánem přežití v řádu několika měsíců, jen výjimečně několika let u alogenně transplantovaných pacientů. Další zlepšování bude vyžadovat detailnější pochopení biologie onemocnění se studiem signálních drah a vývojem nových terapeutických modalit na poli léčby BPDCN.
ZÁVĚR
Nádory z blastických plazmocytoidních dendritických buněk představují vzácnou skupinu hematologických malignit, u nichž blasty vykazují pozitivitu CD4 a CD56. Nízká četnost onemocnění bude vyžadovat vznik mezinárodních registrů a kooperativních skupin, jen tak se dají očekávat další pokroky v diagnostice, klasifikaci a léčbě nemocných s BPDCN. Výsledky prospektivních studií mohou přispět k vytvoření léčebných doporučení, a zlepšit tak zatím obecně nepříznivou prognózu tohoto onemocnění. Současně je nutné zvýšit povědomí o onemocnění, a urychlit tak diagnostiku a zahájení intenzivní terapie nutné ke kontrole průběhu nemoci.
Podíl autorů na rukopise
MČ – hlavní autor práce
TS – léčba nemocného, spoluautor, korespondující autor
ZR – léčba nemocného, kritická revize rukopisu
LR – léčba nemocného, kritická revize rukopisu
MT – diagnostika, kritická revize rukopisu
MD – diagnostika, kritická revize rukopisu
Čestné prohlášení autora
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů, a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Tohle prohlášení se vztahuje i na spoluautory.
Poděkování
Tato práce byla podpořena grantem IGA_LF_2017_007.
Doručeno do redakce dne 21. 5. 2017.
Přijato po recenzi dne 28. 6. 2017.
doc. MUDr. Tomáš Szotkowski, Ph.D.
Hemato-onkologická klinika LF UP a
FN Olomouc
I. P. Pavlova 6
779 00 Olomouc
e-mail: tomas.szotkowski@fnol.cz
Zdroje
1. Pagano L, Valentini CG, Grammatico S, Pulsoni A. Blastic plasmacytoid dendritic cell neoplasm: diagnostic criteria and therapeutical approaches. Br J Haematol 2016;174(2):188–202.
2. Adachi M, Maeda K, Takekawa M, et al. High expression of CD56 (N-CAM) in a patient with cutaneous CD4-positive lymphoma. Am J Hematol 1994;47(4):278–282.
3. Brody JP, Allen S, Schulman P, et al. Acute agranular CD4-positive natural killer cell leukemia. Comprehensive clinicopathologic studies including virologic and in vitro culture with inducing agents. Cancer 1995;75(10):2474–2483.
4. Petrella T, Comeau MR, Maynadié M, et al. ‚Agranular CD4+ CD56+ hematodermic neoplasm‘ (blastic NK-cell lymphoma) originates from a population of CD56+ precursor cells related to plasmacytoid monocytes. Am J Surg Pathol 2002;26(7):852–862.
5. Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009;114:937–951.
6. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016;127:2391–2405.
7. Jacob MC, Chaperot L, Mossuz P, et al. CD4+ CD56+ lineage negative malignancies: a new entity developed from malignant early plasmacytoid dendritic cells. Haematologica 2003;88(8):941–955.
8. Bueno C, Almeida J, Lucio P, et al. Incidence and characteristics of CD4(+)/HLA DRhi dendritic cell malignancies. Haematologica 2004;89(1):58–69.
9. Petrella T, Bagot M, Willemze R, et al. Blastic NK-cell lymphomas (agranular CD4+CD56+ hematodermic neoplasms): a review. Am J Clin Pathol 2005;123(5):662–675.
10. Facchetti F, Cigognetti M, Fisogni S, et al. Neoplasms derived from plasmacytoid dendritic cells. Mod Pathol 2016;29(2):98–111.
11. Jegalian AG, Buxbaum NP, Facchetti F, et al. Blastic plasmacytoid dendritic cell neoplasm in children: diagnostic features and clinical implications. Haematologica 2010;95(11):1873–1879.
12. Pagano L, Valentini CG, Pulsoni A, et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian multicenter study. Haematologica 2013;98(2):239–246.
13. Tsagarakis NJ, Kentrou NA, Papadimitriou KA, et al. Acute lymphoplasmacytoid dendritic cell (DC2) leukemia: results from the Hellenic Dendritic Cell Leukemia Study Group. Leuk Res 2010;34(4):438–446.
14. Julia F, Petrella T, Beylot-Barry M, et al. Blastic plasmacytoid dendritic cell neoplasm: clinical features in 90 patients. Br J Dermatol 2013;169(3):579–586.
15. Leroux D, Mugneret F, Callanan M, et al. CD4(+), CD56(+) DC2 acute leukemia is characterized by recurrent clonal chromosomal changes affecting 6 major targets: a study of 21 cases by the Groupe Français de Cytogénétique Hématologique. Blood 2002;99(11):4154–4159.
16. Lucioni M, Novara F, Fiadrino G, et al. Twenty-one cases of blastic plasmacytoid dendritic cell neoplasm: focus on biallelic locus 9p21.3 deletion. Blood 2011;118:4591–4594.
17. Taylor J, Kim SS, Stevenson KE, et al. Loss-of-function mutations in the splicing factor ZRSR2 are common in blastic plasmacytoid dendritic cell neoplasm and have male predominance. Blood 2013;122:741.
18. Reimer P, Rüdiger T, Kraemer D, et al. sWhat is CD4+CD56+ malignancy and how should it be treated? Bone Marrow Transplant 2003;32(7):637– 646.
19. Aoki T, Suzuki R, Kuwatsuka Y, et al. Long-term survival following autologous and allogeneic stem cell transplantation for blastic plasmacytoid dendritic cell neoplasm. Blood 2015;125(23):3559–3562.
20. Dalle S, Beylot-Barry M, Bagot M, et al. Blastic plasmacytoid dendritic cell neoplasm: is transplantation the treatment of choice? Br J Dermatol 2010;162(1):74–79.
21. Poret E, Vidal Ch, Desbrosses Y, et al. How to treat blastic plasmacytoid dendritic cell neoplasm (BPDCN) patients: results on 86 patients of the French BPDCN Network. Blood 2015;126:456.
22. Pemmaraju N, Thomas D, Kantarjian HM, et al. Analysis of Outcomes of Patients with Blastic Plasmacytoid Dendritic Cell Neoplasm. Blood 2012;120:3554.
23. Kharfan-Dabaja MA, Lazarus HM, Nishihori T, Mahfouz RA, Hamadani M. Diagnostic and therapeutic advances in blastic plasmacytoid dendritic cell neoplasm: a focus on hematopoietic cell transplantation. Biol Blood Marrow Transplant 2013;19(7):1006–1012.
24. Frankel AE, Woo JH, Ahn C, et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood 2014;124:385–392.
25. Stein EM, Tallman MS. Emerging therapeutic drugs for AML. Blood 2016;127:71–78.
26. Pevná M, Kissová J, Doubek M, Adam Z, Klabusay M, et al. Leukemie z dendritických buněk CD4+56+, typ DC2. Vnitř Lék 2010;56(Suppl 2):183–187.
27. Klabusay M, Pevná M, Kissová J, et al. Vzácná diagnóza: CD4+56+ leukémie z dendritických buněk typu DC2. Čas Lék Čes 2008;147:511–515.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2017 Číslo 3
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- Klinický význam menších rozestupů mezi silami přípravků s levothyroxinem v léčbě hypotyreózy
- Umíme správně řešit osteporotické zlomeniny?
- Antikoagulační léčba u pacientů před operačními výkony
- Prof. Miloš Broďák: Zavedení nových preparátů výrazně zlepšilo výsledky i u progredujících a recidivujících karcinomů prostaty
Nejčtenější v tomto čísle
- Doporučení ČHS pro diagnostiku a léčbu imunitní trombocytopenie (ITP)
- Dlouhodobé výsledky léčby chronické myelomonocytární leukemie ve vybraných hematologických centrech
- Nádor z blastických plazmocytoidních dendritických buněk: kazuistika a přehled literatury
- Získaný angioedém s deficitem C1 inhibitoru u pacientky s B-lymfomem nízké malignity a efekt léčby základního onemocnění na projevy angioedému