
Molekulární mechanizmy primární a sekundární rezistence, molekulárně-genetické znaky a vlastnosti KIT/PDGFRA nemutovaných GIST
Molecular mechanisms of primary and secondary resistance, molecular-genetic features and characteristics of KIT/PDGFRA non-mutated GISTs
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract. Most of them arise due to activating mutations in KIT (75 – 85 %) or PDGFRA (less than 10 %) genes. Identification of the activating mutations in KIT and PDGFRA genes, which code for receptor tyrosine kinases (RTKs), has improved the outcome of targeted therapy of metastatic, unresectable or recurrent GISTs. Primary and/or secondary resistance represents a significant problem in the targeted therapy by Imatinib mesylate (IM) in patients with GIST. An important mechanism of the secondary resistance is the evolvement of secondary mutations. Except for primary and secondary resistance, there is another problem of disease progression - a failure of tumor cells eradication even in the long term therapy of tyrosine kinase inhibitors. GISTs without mutations in KIT/PDGFRA genes constitute 10 - 15% GISTs in adults, and a majority (85 %) of pediatric GISTs. KIT/PDGFRA wild-type GISTs represent a heterogeneous group of tumors with several molecular-genetics and/or morphologic differences. KIT/PDGFRA wild-type GISTs are different in their molecular features, for example in mutations in the BRAF, KRAS, NF1 genes or defects of succinate dehydrogenase (SDH) subunits. KIT/PDGFRA wild-type GISTs are generally less sensitive to targeted therapy by tyrosine kinase inhibitors in comparison with KIT/PDGFRA mutated GISTs. Inhibitors of BRAF, PI3K (mTOR) or inhibitors of IGF1R and VEGFR receptors provide alternative therapeutic strategies.
Keywords:
GIST – primary and secondary resistance – KIT/PDGFRA non mutated GISTs – SDH complex – BRAF mutations – IGH system
Autoři:
Alena Kalfusová; Roman Kodet
Působiště autorů:
Ústav patologie a molekulární medicíny, 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Čes.-slov. Patol., 53, 2017, No. 4, p. 167-173
Kategorie:
Přehledový článek
Souhrn
Gastrointestinální stromální nádory (GIST) tvoří nejčastější mezenchymální nádory gastrointestinálního traktu. Velká část z nich vzniká na základě aktivačních mutací v genech KIT (75 - 85 %) nebo PDGFRA (méně než 10 %). Identifikace aktivačních mutací v genech KIT a PDGFRA, které kódují příslušné receptorové tyrozinkinázy (RTK), znamenala významný průlom v cílené terapii metastatických, neoperabilních nebo recidivujících GIST. Primární a/nebo sekundární rezistence je významným problémem biologické terapie imatinib mesylátem (IM) u pacientů s GIST. Oba typy rezistence vedou ke snížené terapeutické odpovědi na IM, případně k selhání této léčby a progresi onemocnění. Hlavním mechanizmem sekundární rezistence je vznik sekundárních mutací. Problémem progrese onemocnění je kromě primární a sekundární rezistence také selhání úplné eradikace nádorových buněk i při dlouhotrvající terapii inhibitory RTK. GIST bez přítomnosti mutací v genech KIT/PDGFRA tvoří u dospělých 10 - 15 %, u pediatrických GIST je to až 85 % případů. KIT/PDGFRA nemutované GIST tvoří heterogenní skupinu nádorů s několika molekulárně-genetickými i morfologickými odlišnostmi. Molekulárně se KIT/PDGFRA nemutované GIST liší např. mutacemi v genech BRAF, KRAS, NF1 nebo defekty v sukcinát dehydrogenázových (SDH) podjednotkách. GIST s nemutovanými geny KIT/PDGFRA vykazují nižší citlivost na cílenou terapii IM v porovnání s mutovanými GIST. Terapeutické možnosti se ve skupině KIT/PDGFRA nemutovaných GIST soustřeďují na BRAF inhibitory, inhibitory PI3K dráhy (mTOR), nebo na inhibitory receptorů IGF1R a VEGFR.
Klíčová slova:
GIST – primární a sekundární rezistence – KIT/PDGFRA nemutované GIST – SDH komplex – BRAF mutace – IGH systém
Většina gastrointestinálních stromálních nádorů (GIST) vzniká na podkladě aktivačních mutací v genech KIT (75 - 85 %) nebo PDGFRA (méně než 10 %). Mutace jsou navzájem exkluzivní a často heterozygotní. Ve vzácných případech familiárních GIST se onemocnění rozvine na základě vrozených autozomálně dominantních mutací v genech KIT nebo PDGFRA. Mutované GIST mají podobné charakteristiky. Morfologicky jsou tvořeny vřetenobuněčnými nebo epiteloidními buňkami. V zastoupení pohlaví nejsou patrné rozdíly. Tyto GIST postihují pacienty nad 50 let věku a mohou se vyskytovat kdekoliv v rámci gastrointestinálního traktu, méně často v omentu, mesenteriu, retroperitoneu nebo v oblasti malé pánve (1). Souvislost anatomické lokalizace nádoru s přítomnosti některých mutací v genech KIT a PDGFRA můžeme vysvětlit rozdílným původem intersticiálních Cajalových buněk (Interstitial cells of Cajal, ICC). Nádory s mutacemi v exonu 9 genu KIT se kupříkladu přednostně vyskytují v tenkém střevě. Tím se liší od nádorů s mutacemi v exonu 18 genu PDGFRA (D842V), které se nacházejí pouze v žaludku, mesenteriu a v omentu. Důvodem je, že nádory s mutacemi v exonu 9 genu KIT vznikají z odlišné podskupiny ICC buněk. Mutace v exonu 11 genu KIT, které se vyskytují nejčastěji, nacházíme naproti tomu v průběhu celého gastrointestinálního traktu (GIT). Možným vysvětlením je, že pocházejí z tzv. ubikvitního podtypu ICC buněk (2).
PRIMÁRNÍ A SEKUNDÁRNÍ REZISTENCE
Největším průlomem v terapii GIST byl objev aktivačních mutací v genu KIT (3) a v genu PDGFRA (4), které kódují příslušné receptorové tyrozinkinázy (RTK). Spojení těchto poznatků s poznatky úspěšné terapie chronické myeloidní leukemie (CML), která je cílená na fúzní protein BCR-ABL, rovněž s tyrozinkinázovou aktivitou (5), odstartovalo významnou kapitolu v léčbě metastatických, neoperabilních nebo recidivujících GIST. Terapie je u obou klinických jednotek založená na inhibici kinázové aktivity vazbou inhibitoru (imatinib mesylátu, IM) na ATP vazebné místo proteinu. V průběhu terapie se však u obou typů onemocnění setkáváme se vznikem sekundární rezistence. U CML je to přítomnost sekundárních mutací v kinázových doménách BCR-ABL fúzního proteinu nebo amplifikace genu BCR-ABL (6,7). U pacientů s GIST se setkáváme s primární a sekundární rezistencí (8). V domácím písemnictví jsme na problematiku primární a sekundární rezistence GIST s progresí na terapii ve stručném přehledu již upozornili (9). Otázka získané rezistence a s ní spojeného selhávání léčby je nadále aktuálním a důležitým tématem pro řešení volby cílené biologické léčby.
„Časná“ nebo také „primární“ rezistence je definována jako progrese onemocnění v průběhu prvních 3 - 6 měsíců od začátku terapie IM. Týká se přibližně 15 - 20 % pacientů. Hlavním důvodem vzniku primární rezistence je mutační stav genů KIT nebo PDGFRA (typ mutace a/nebo lokalizace primární mutace)(1,10). S primární rezistencí nejčastěji souvisí mutace v exonu 9 genu KIT, mutace v genu PDGFRA (zejména mutace D842V v exonu 18), nebo naopak absence mutací jak v genu KIT, tak genu PDGFRA (10). Exon 9 genu KIT kóduje extracelulární doménu, která za normálních okolností inhibuje dimerizaci receptoru KIT. Mutace v exonu 9 způsobí sterickou překážku, která nedovolí IM pevně se navázat na katalytickou doménu receptoru (TK1 doména). V případě genu PDGFRA exon 18 kóduje aktivační „smyčku“ (TK2 doména). Bodová mutace D842V v exonu 18 vede k substituční záměně na tomto místě a ke vzniku stabilně aktivní konformace proteinu. Uvedená aktivní konformace receptoru na vazbu IM a jeho inhibiční vlastnosti neodpovídá.
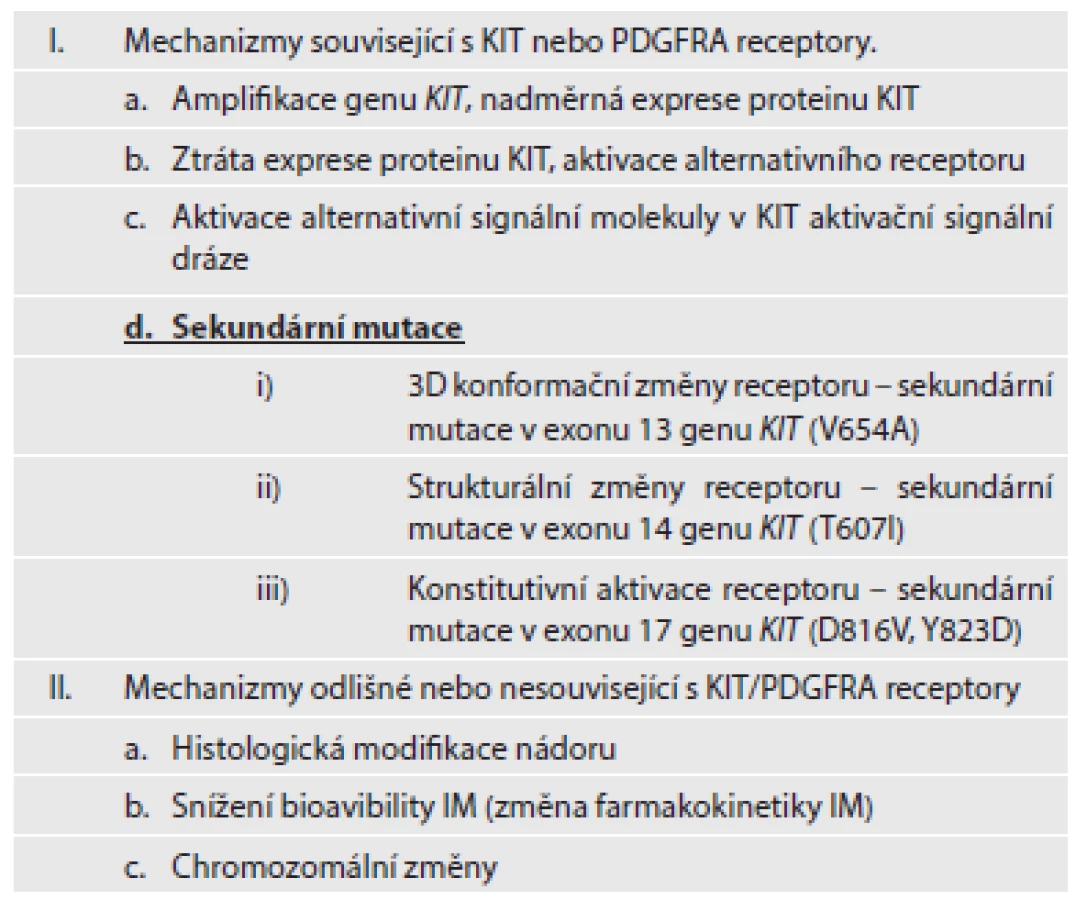
„Získaná“ nebo-li „sekundární“ rezistence vzniká v průběhu terapie u pacientů, jejichž GIST prvotně na terapii odpovídal, případně u nich došlo ke stabilizaci onemocnění, ovšem v průběhu několika měsíců na terapii nereagují a dochází u nich k progresi onemocnění. Časové rozmezí vzniku sekundární rezistence se pohybuje v rozpětí 6 až 24 měsíců od začátku terapie IM. Hlavním mechanizmem vzniku získané rezistence je přítomnost sekundárních mutací. Sekundární mutace se vyskytují u 50 - 70 % pacientů s progresí onemocnění. Přítomnost sekundárních mutací zjišťujeme u pacientů, kteří měli detekovány primární mutace v genu KIT, v menší míře také u těch, kteří měli primární mutace lokalizovány v genu PDGFRA. Téměř výhradně se sekundární mutace vyskytují ve stejném genu a na stejné alele jako primární mutace (1). Sekundární mutace se shlukují ve dvou oddělených KIT tyrozinkinázových doménách (TK1 a TK2). První je ATP/imatinib vazebné místo (exony 13 a 14 genu KIT), druhá je kinázová aktivační smyčka (exony 17 a 18 genu KIT)(11). Sekundární mutace ATP vazebného místa přímo inhibují vazbu IM. Sekundární mutace V654A (exon 13 genu KIT) vede ke snížení vazebné aktivity IM. Sekundární mutace v exonu 14 genu KIT, tj. T607I, je často označovaná jako „strážce brány“ – gatekeeper. Tato mutace je zodpovědná za tvorbu sterické překážky pro vazbu IM (12). Mutace T670I je homologní s mutaci T315I u CML (fúzního genu BCR-ABL), která způsobuje silnou rezistenci, a s mutací T790M u genu EGFR (13). Mutace v aktivační smyčce (exon 17 genu KIT) destabilizují inaktivní formu RTK (protein KIT je konstitutivně aktivován)(12). Přítomnost sekundárních mutací doposud nebyla popsána u pacientů, kteří nepodstoupili terapii IM (14). Stručný přehled mutací zodpovědných za sekundární rezistenci poskytuje tabulka č. 1.

Nepříznivým znakem sekundární rezistence je také velká heterogenita sekundárních (rezistentních) mutací. Sekundární mutace se mohou u jednoho pacienta lišit co do počtu nebo typu jednak v rámci jednoho nádorového ložiska, či metastázy, ale mohou se lišit i mezi jednotlivými metastázami (1). Rozdílné sekundární mutace v rozdílných metastázách u stejného pacienta poukazují na rozdílnou klonální evoluci (15). Heterogenita rezistentních (sekundárních) mutací „v“ jednotlivé metastáze a „mezi“ metastázami u jednotlivých pacientů může být značná. Může kolísat od dvou do pěti sekundárních mutací v rozdílných metastázách nebo i vznikem dvou rozdílných mutací v jedné metastáze (16,17). Variabilita množství i lokalizace sekundárních mutací (u jednoho pacienta mohou být přítomny mutace v exonu 13 a rovněž i v exonu 17 genu KIT) je závažným problémem pro cílenou biologickou léčbu s ohledem na jejich rozdílnou senzitivitu. Molekulárně-genetická analýza provedená z jednoho metastatického ložiska tak nemusí představovat skutečný mutační stav nádoru. Sekundární mutace v KIT kinázových doménách nebyly detekovány u nemutovaných genů KIT/PDGFRA, u mutovaných GIST s nezvyklou morfologií a/nebo u GIST se ztrátou exprese KIT proteinu (16).
Mezi další mechanizmy sekundární (získané) rezistence patří amplifikace genu KIT a/nebo PDGFRA a následující nadměrná exprese daných proteinů. Aktivace alternativních RTK (např. AXL) je jedním z dalších mechanizmů získané rezistence. Mimo uvedené mechanizmy změny struktury nebo funkce receptorů KIT a PDGFRA, patří k dalším mechanizmům získané rezistence chromozomální modifikace, změny ve farmakokinetice IM, popřípadě diferenciace nádoru (18). Stručný přehled mechanizmů sekundární rezistence je uvedený v tabulce č. 2.
Problémem progrese onemocnění je kromě primární a sekundární rezistence také selhání úplné eradikace nádorových buněk, popřípadě kmenových buněk, které jsou jejich prekurzory v nádorovém ložisku. Kompletní odstranění nádorových buněk selhává i při dlouhotrvající terapii inhibitory RTK. Inhibitory RTK selhávají při odstranění nádorových buněk v 95 - 97 % případů. Rozpětí redukovaných nádorových buněk může kolísat od méně než 10 % až po více než 90 %. Absolutního odstranění nádorových buněk ovšem nebylo dosaženo nikdy (1,2). Přežívající nádorové buňky jsou metabolicky v klidovém stadiu a neproliferují. Jejich výstup z buněčného cyklu je však reverzibilní (1). Přetrvávání nádorových buněk navzdory pokračující terapii pomocí inhibitorů RTK umožňuje selekci klonů se sekundárními, na terapii rezistentními mutacemi. Přežívání nádorových buněk i u imatinib-senzitivních mutací je umožněno existencí skupiny buněk s nízkou expresí proteinu KIT, nebo buněk, u kterých vznikla ztráta exprese proteinu KIT (KIT-low/KIT-). Ačkoliv inhibitory RTK mohou kontrolovat diferenciaci, přežívání či proliferaci KIT+ buněk, nemohou odstranit imatinib-rezistentní KIT-low/KIT- kmenové buňky. Tyto buňky se následně stávají zdrojem relapsu onemocnění. Vznik sekundárních mutací rezistentních na IM v přežívajících prekurzorech může umožnit jejich diferenciaci v KIT+ buňky a jejich nekontrolovatelný růst. Předpokládaný model procesu přetrvávání nádorových buněk a klonální selekci buněk se získanými mutacemi uvádíme na obr. 1.

Modré kroužky KIT-: KIT negativní kmenové buňky (nebo jejich prekurzory), nesou KIT mutace senzitivní na imatinib, exprese proteinu KIT je ale velmi slabá nebo není přítomná vůbec (KIT-low/KIT-). Modré kroužky KIT+: KIT pozitivní buňky, vznikající z KIT-low/KIT- (s přítomnými mutacemi senzitivními na imatinib). Prázdné, bílé kroužky: představují mrtvé buňky. Světle růžové kroužky: KIT-low/KIT- kmenové buňky nebo jejich prekurzory, které obsahují sekundární mutace rezistentní na imatinib. Tmavě růžové kroužky: KIT+ buňky vznikající z KIT-low/KIT- prekurzorů se sekundárními mutacemi rezistentními na imatinib. Modré šipky: terapie imatinibem. Bílá šipka: ukončení terapie imatinibem.
Po morfologické stránce můžeme u nádorů, které jsou terapeuticky ovlivněny pozorovat změnu diferenciace nádorových buněk v jiný buněčný typ. Buňky se mohou transformovat v rabdomyoblastický, chondroidní, nebo kostní typ. Mechanizmus transformace můžeme vysvětlit snahou nádorových buněk (pod vlivem terapeutického tlaku) vyhnout se apoptóze. Programovanou smrt buňky obejdou tyto buňky opuštěním buněčného cyklu a spustí expresi genů, které jsou spojené s jiným diferencovaným typem buněk. Nevyjasněnou otázkou zatím zůstává průběh této transformace. Zdrojem mohou být buňky, které se po přerušení terapie vyhnuly apoptóze a opět proliferují, nebo jsou jejich zdrojem nádorové kmenové buňky. Ve vzácných případech se může GIST transformovat do high grade anaplastického sarkomu bez exprese CD117 povrchového proteinu (2). Tato změna byla pozorována u nádorů bez terapie, ale rovněž i u nádorů léčených (19). Rabdomyoblastické buňky u rezistentních GIST s progresí onemocnění exprimují typicky desmin a myogenin (20,21).
KIT/PDGFRA NEMUTOVANÉ GIST
Donedávna byl z terapeuticko-indikačního hlediska nejvýznamnější a nejčastěji sledován mutační stav genů KIT a PDGFRA. Sledování přítomnosti a lokalizace primárních mutací, výskytu sekundárních mutací při progresi onemocnění, a s tím spojené primární a/nebo získané rezistence na cílenou terapii IM, byly dlouhodobě důležitým cílem molekulárně-genetické analýzy GIST. V posledních letech se zajímavou a z terapeutického hlediska otevřenou kapitolou stávají GIST, u kterých nebyly zjištěny mutace v těchto genech.
GIST bez přítomnosti mutací v genech KIT/PDGFRA tvoří u dospělých 10 - 15 % případů, u mnohem vzácnějších pediatrických GIST je to až 85 % případů. Podle množství doposud publikovaných studií se u tzv. wild-type (KIT/PDGFRA nemutovaných) GIST jedná o komplex několika podtypů. Tyto podtypy GIST mají rozdílné molekulární znaky. Jsou jimi např. mutace v genech BRAF, KRAS, NF1 nebo defekty v sukcinát dehydrogenázových (SDH) podjednotkách (22). Rozdělení a charakteristice KIT/PDGFRA nemutovaných GIST podle jejich molekulárních vlastností se mimo zahraniční literaturu podrobně věnovala i domácí publikace (23). Pro úplnost a zpřehlednění problematiky heterogenity KIT/PDGFRA nemutovaných GIST slouží obr. 2.

Podle imunohistochemické (IHC) exprese SDHB podjednotky dělíme KIT/PDGFRA nemutované GIST do dvou skupin, na SDHB+ (zahrnuje GIST s mutacemi v genu NF1 a sporadické GIST s nebo bez přítomnosti mutací v genech BRAF nebo KRAS) a na SDHB- (nebo také s defektem SDH komplexu). Skupinu SDHB- KIT/PDGFRA nemutovaných GIST podle IHC exprese SDHA podjednotky dále dělíme na SDHA+ (SDHB-/SDHA+), kam patří GIST souvisící s Carney-Stratakis syndromem (vrozené nebo somatické inaktivační mutace v genech SDHB, SDHC, nebo SDHD), GIST jako součást Carneyovy triády (bez přítomnosti mutací v genech SDH komplexu) a sporadické GIST s vrozenými nebo somatickými mutacemi v genech SDH komplexu. SDHB-/SDHA- GIST jsou charakterizovány vrozenými nebo somatickými inaktivačními mutacemi v genu SDHA (mladí dospělí/ pediatrický typ GIST).
V krátkosti lze uvést, že KIT/PDGFRA nemutované GIST můžeme podle imunohistochemické (IHC) exprese SDHB podjednotky rozdělit do dvou skupin. Do první skupiny, „typ 1“, patří GIST s expresí SDHB podjednotky (SDHB pozitivní, SDHB+). Druhou skupinu, „typ 2“, tvoří SDHB negativní (SDHB-) GIST. Do první skupiny spadají GIST se somatickou inaktivací genu NF1 (ztráta heterozygozity nemutované NF1 alely) a některé sporadické GIST. Jak vyplývá z charakteristiky těchto GIST nemají sporadické GIST z této skupiny mutace v genech KIT/PDGFRA. Přibližně v 15 % případů však mohou mít mutovány geny BRAF nebo KRAS. GIST s mutacemi v genu NF1 se běžně vyskytují v tenkém střevě s typicky multifokálním výskytem nádoru a neexprimují protein IGF1R.
Druhou skupinu GIST SDHB- můžeme rozdělit podle IHC exprese SDHA podjednotky (exprese je ovlivněna inaktivačními mutacemi genu SDHA) na SDHA pozitivní (SDHA+) a SDHA negativní (SDHA-). Do skupiny SDHA pozitivních GIST patří GIST jako součásti Carneyovy triády, syndromu Carney-Strakatis, a sporadické GIST. Tyto sporadické GIST opět nemají mutovány geny KIT/PDGFRA, mají ovšem mutované geny pro SDH komplex (SDHB, SDHC, SDHD, méně obvykle SDHA). GIST jsou v těchto případech běžně lokalizovány v žaludku a je u nich známa menší prevalence ženského pohlaví. Histologicky se ovšem podobají GIST bez exprese proteinu SDHA. Ve skupině SDHA negativních (SDHA-) GIST jsou nádory, které jsou diagnostikovány u mladých dospělých (mladých žen). Charakteristická je lokalizace v žaludku, s typickými morfologickými a fenotypovými znaky: smíšeným epiteloidním nebo vřetenobuněčným vzhledem, difúzní pozitivitou povrchových molekul KIT a DOG1 a nadměrnou expresí proteinu IGF1R. Častý je u nich výskyt metastáz v lymfatických uzlinách. Navzdory výskytu metastáz je u těchto pacientů průběh nemoci indolentní (1,22).
KIT/PDGFRA nemutované GIST s mutacemi v genech BRAF/KRAS/PI3K
Protein BRAF patří k serin/treoninovým proteinkinázam a jeho funkce spočívá v kontrole proliferace a diferenciace buněk prostřednictvím RAS-RAF-MAPK signální dráhy. Protein BRAF je kódovaný stejnojmenným genem BRAF (24). S mutovaným genem BRAF se můžeme setkat u širokého spektra nádorů od benigních melanocytárních névů, hyperplastických polypů v tenkém střevě, low-grade gliomů až po melanomy, vlasatobuněčnou leukemii (hairy-cell leukemia, HCL), papilární karcinomy štítné žlázy a kolorektální karcinomy (25). Přítomnost mutací v genu BRAF byla detekovaná i u GIST (20). Zastoupení mutací v genu BRAF je u každé z těchto klinických jednotek rozdílné. U HCL je mutovaných případů až 100 %. V případě dalších klinických jednotek je procentuální zastoupení nižší (např. melanom 50 %, papilární karcinom štítné žlázy 45 %, low-grade gliomy 35 - 40 %, kolorektální karcinom 11 %). U GIST se počet BRAF mutovaných případů pohybuje v rozpětí 3 - 13 %.
Nejčastěji jsou mutace v genu BRAF lokalizovány v exonu 15, který kóduje kinázovou doménu proteinu. Ve většině případů se jedná o jednoduchou substituci v kodonu 600, která vede k záměně valinu za kyselinu glutamovou (V600E). Méně často se mutace v genu BRAF vyskytují v exonu 11. GIST s mutacemi v genu BRAF jsou morfologicky nerozlišitelné od GIST s mutacemi v genech KIT/PDGFRA. Přednostně se vyskytují v tenkém střevu a u ženského pohlaví (26). Prvotní analýzy ukazovaly, že mutace v genech KIT/PDGFRA jsou s mutacemi v genu BRAF mutačně exkluzivní (20,24). Následně byl ovšem prokázán současný výskyt mutací v genech KIT nebo PDGFRA a BRAF u GIST i u melanomů.
Přítomnost mutací v genu BRAF u KIT/PDGFRA nemutovaných GIST ozřejmila patomechanizmus vzniku této skupiny GIST. Mutace v genu BRAF u GIST vedou ke vzniku a predikují primární rezistenci na cílenou terapii IM. V experimentech na buněčných liniích se současnou expresí KIT senzitivních mutací a konstitutivně aktivovaných proteinů KRAS nebo BRAF bylo prokázáno, že cílená terapie IM inhibuje protein KIT. Neovlivní ovšem intracelulární signální dráhu aktivovanou prostřednictvím mutovaných genů BRAF/KRAS (27). Kromě primární rezistence se mutace v genu BRAF podílejí na vzniku sekundární rezistence. Mutace v genu BRAF byly zjištěny u nádorů s progresí na terapii, které prvotně odpovídaly na terapii a neměly sekundární mutace v genech KIT nebo PDGFRA (20).
Mutace v genu pro fosfatidyl inositol 3 kinázu (PI3K) byly rovněž prokázány u rezistentních GIST s progresí na terapii. Protein PI3K je součásti aktivační dráhy RTK KIT a PDGFRA. Reguluje buněčný růst, proliferaci a přežívání buněk. Pacienti s mutacemi v genech BRAF nebo PI3K (ovlivňující primární nebo sekundární rezistenci u GIST) mohou profitovat z alternativních terapeutických postupů (BRAF inhibitory nebo inhibitory PI3K signální dráhy, kterými jsou inhibitory p110α nebo mTOR) (28). Na rozdíl od mutací v genu BRAF se mutace v genech ze skupiny RAS (NRAS, KRAS) vyskytují u GIST ojediněle (20,26,29,30).
KIT/PDGFRA nemutované GIST a SDH komplex
SDH (sukcinát dehydrogenázový) komplex tvoří 4 proteinové podjednotky: SDHA, SDHB, SDHC, a SDHD, které jsou kódovány příslušnými geny. SDH komplex je lokalizovaný na vnitřní membráně mitochondrií a je nepostradatelný pro buněčný metabolizmus. Přítomnost inaktivačních mutací nebo poškození některé z části SDH komplexu (SDHA, SDHB, SDHC, nebo SDHD) způsobí ztrátu proteinové exprese podjednotky SDHB (31). Zatímco ztráta proteinové exprese SDHB může být ovlivněna defektem v kterékoliv z podjednotek SDH komplexu, proteinová exprese podjednotky SDHA (pozitivita/negativita) je ovlivněna změnou pouze v genu SDHA. Ztráta proteinové exprese podjednotky SDHA je specificky spojena s přítomnosti inaktivačních mutací v genu SDHA.
Změny v SDH podjednotkách jsou z jedné poloviny případů ovlivněny přítomnosti mutací v kterémkoliv z SDHA, SDHB, SDHC, nebo SDHD genů. Druhá polovina mutacemi v SDH komplexu ovlivněna není. Možným patomechanizmem vzniku této skupiny poruch je epigenetické utlumení aktivity SDH komplexu. V porovnání s jinými GIST byla u GIST s defekty SDH komplexu pozorována jejich značná hypermetylace (32). Mutace v SDH komplexu vedou k poruše funkce mitochondriálního komplexu II (v řetězci transportu elektronů) a následně k narušení oxidativní fosforylace. SDH komplex oxiduje sukcinát na fumarát. Porucha oxidativní fosforylace vede k hromadění sukcinátu a vzniká tak efekt pseudohypoxie prostřednictvím nadměrné exprese proteinů HIF (hypoxia inducible factor) (32). Sukcinát inhibuje degradaci HIF1α, což následně vede ke zvýšení jeho hladin a translokaci v jádře. V jádře pak spouští transkripci růstových faktorů IGF1R a VEGFR (33,34). Zárodečné mutace v genech SDHB, SDHC a SDHD vedou ke vzniku syndromu Carney-Strakatis s familiárním výskytem paragangliomu a GIST.
U sporadických GIST s nemutovanými geny KIT/PDGFRA byly popsány somatické mutace v genech SDH komplexu. Zahraniční studie popisují přítomnost zárodečných mutací i u sporadických GIST u dospělých bez zjevné souvislosti s familiárním výskytem nebo paragangliomem. Pravděpodobným vysvětlením je, že se jedná o utlumenou formu Carney-Strakatis syndromu nebo o nejednoznačnou manifestaci či definici tohoto syndromu (35). Carneyova triáda (nefamiliární výskyt paragangliomů, GIST a plicních chondrohamartomů) je rovněž definována poruchou v SDH komplexu, ale mutace v genech SDHA, SDHB, SDHC, nebo SDHD nebyly u této klinické jednotky zatím popsány (36).
GIST s defektem v některé z SDH podjednotek se vyznačují epiteloidní morfologii, mnohočetným výskytem, rezistencí na terapii IM a charakteristickou lokalizací v žaludku. Ve studii čítající více než 1000 dobře definovaných GIST s defektem SDH komplexu byly všechny v žaludku (37). Na rozdíl od „klasických“ GIST, tyto GIST metastazují do lymfatických uzlin a vykazují expresi proteinu IGF1R. Jejich prognóza se nedá stanovit jen na základě velikosti nádoru, lokalizace a mitotické aktivity. Pacienti mohou i při rozvoji jaterních metastáz dlouhodobě přežívat (34).
KIT/PDGFRA nemutované GIST a IGF1R
IGF (insulin-like growth factor) systém tvoří IGF ligandy (IGF1 a IGF2), receptory (IGFR a insulinový receptor), a 6 insulin-vazebných proteinů (IGFBP1-6). Všechny tyto složky IGF systému hrají významnou roli v růstu a vývinu mnoha tkání a regulují celkový buněčný růst (33). Receptor pro růstový faktor podobný insulinu, typ 1 (insuline-like growth factor receptor 1, IGF1R), je podobně jako RTK KIT a PDGFRA, transmembránový receptor s tyrosinkinázovou aktivitou. Aktivuje signální dráhu PI3K nebo MAPK. Bylo prokázáno, že IGF1R ve spolupráci s ligandy IGF1 a IGF2 se uplatňuje v transformaci několika onkogenů. Typickým, a podle všeho také specifickým znakem GIST s defektem SDH komplexu je nadměrná exprese proteinu IGF1R (32,35,37,38). Možným mechanizmem nadměrné exprese IGF1R u GIST s defektem SDH komplexu je aktivace prostřednictvím nadměrné exprese HIF1α faktoru (33). Aktivace IGF1R signální dráhy je možným alternativním terapeutickým cílem u GIST s defektem SDH komplexu (35).
KIT/PDGFRA nemutované GIST a NF1
Neurofibromatóza typu 1 (NF1) (von Reclinghausenova choroba) je autozomálně dominantní dědičné onemocnění, charakterizované přítomností mnohočetných neurofibromů, skvrn typu „café-au-lait“ a dalšími mezenchymálními nádory. NF1 vzniká jako následek inaktivačních mutací v genu NF1, který kóduje protein neurofibromin. Neurofibromin je analogem GTP aktivačních proteinů (GAP), které regulují aktivitu proteinů RAS (34,39). Přibližně u 7 % pacientů s NF1 se rozvine GIST. Početnost nádorů kolísá od jednoho ložiska po jejich mnohočetný výskyt. Nejčastější lokalizací je tenké střevo. Velká část těchto GIST má nemutované geny KIT a PDGFRA, ale podle očekávání mají buď somatické mutace v genu NF1 nebo ztrátu nemutované NF1 alely. Výsledkem obou molekulárních změn je aktivace signálních drah prostřednictvím MAP kinázové dráhy (2). Často jsou tyto GIST nízkého rizika s klinicky indolentním průběhem (40).
PEDIATRICKÉ GIST
Pediatrické GIST mají mutace v genech KIT a/nebo PDGFRA pouze v 15 - 20 %. S tím souvisí i horší odpověď dětských pacientů na cílenou terapii inhibitorem RTK. Pediatrické GIST a GIST s nemutovanými geny KIT/PDGFRA vykazují ztrátu proteinové exprese podjednotky SDHB (10). Z celkového počtu GIST v populaci tvoří dětské GIST (častěji u mladých dívek) přibližně jen 1 - 2 %. Přes absenci mutací v genech KIT/PDGFRA u většiny pacientů exprimují tyto GIST protein KIT stejně tak jako dospělí pacienti (13,40). Pediatrické GIST podobně jako GIST v souvislosti se syndromem Carney-Stratakis nebo s Carneyovou triádou vykazují podobné charakteristiky (33). Prakticky identické znaky jako u pediatrických GIST můžeme najít i u příležitostně se vyskytujících dospělých GIST tzv. „pediatrického typu“. Pravděpodobným vysvětlením je právě společný defekt v SDH komplexu (33).
Pediatrické GIST se vyskytují vzácně, postihují adolescentní dívky (medián věku 14 let), vyznačují se multifokálním výskytem, epiteloidní morfologií, lokalizací nádoru v žaludku (více než v 90 %), metastázami v lymfatických uzlinách a indolentním chováním (41).
TERAPEUTICKÉ MOŽNOSTI
V algoritmu cílené léčby u metastatických nebo recidivujících GIST s mutacemi v genech KIT/PDGFRA se v první linii terapie dosud úspěšně používá imatinib mesylát (IM). Pacienti se vzniklou rezistencí (primární nebo sekundární) na IM jsou ve druhé linii léčeni sunitinib malátem (SM). SM silně inhibuje mutace vzniklé v ATP vazebním místě. Nicméně, SM je méně účinný v inhibici mutací přítomných v aktivační smyčce (exon 17, gen KIT), které tvoří 50 % imatinib-rezistentních mutací (42). Ve třetí linii léčby je u pacientů s metastatickým nebo recidivujícím GIST, kteří neodpovídají na léčbu IM nebo SM schválen regorafenib. Regorafenib je dalším tyrosinkinázovým inhibitorem (TKI), který přímo inhibuje protein KIT. Kromě toho inhibuje signální molekuly RAS, BRAF v dráze aktivace přes RTK PDGFRB (43). Navíc, na rozdíl od IM, regorafenib inhibuje endotelové buňky prostřednictvím VEGFR-2 (vascular endothelial growth factor receptor 2). VEGFR-2 má rozhodující význam v normální i nádorové neovaskularizaci. Regorafenib tak působí na několika úrovních inhibice růstu nebo proliferace buněk. Je rovněž možné, že regorafenib inhibuje i další signální dráhy (např. fibroblast growth factor receptor 1), což může prostřednictvím doposud neznámého kompenzačního mechanizmu přispívat k rezistenci GIST (44).
Účinnost nových TKI, např. ponatinibu (NTC 01874665) se zkouší u pacientů, kteří neodpovídají ani na terapii regorafenibem (43). Monoterapie IM, SM, nebo regorafenibem neumožňuje obsáhnout celou šíří sekundárních mutací (přítomnost několika typů mutací v jednom ložisku) v ATP vazebním místě a stejně tak v aktivační smyčce (42). Potenciál ponatinibu vůči předchozí terapii I., II. a III. linie je podpořen inhibicí GIST se sekundárními mutacemi v exonu 17 genu KIT.
Zajímavou kapitolu v terapii GIST tvoří pacienti s nemutovanými geny KIT/PDGFRA. V porovnání s KIT/PDGFRA mutovanými GIST, je odpověď na terapii IM u nemutovaných GIST téměř o polovinu nižší (44 - 45 % vs. 70 - 71 %)(31). Nemutované GIST představují heterogenní skupinu klinických jednotek s rozdílným patomechanizmem vzniku a odlišnou citlivostí na cílenou terapii. Příkladem jsou gastrické GIST s negativitou exprese proteinu SDHB a s vysokou expresí IGF1R. Prvotně neodpovídají na terapii IM, odpovídají ovšem na terapii SM.
Další možnosti terapie pomocí inhibitorů BRAF, nebo inhibitorů PI3K signální dráhy (inhibitory p110α a/nebo mTOR) se využívají ve skupině KIT/PDGFRA nemutovaných GIST s aktivaci BRAF nebo PI3K signální dráhy. U KIT/PDGFRA nemutovaných GIST je slibnou alternativní cestou léčby inhibice receptoru IGF1R. Aktuálně probíhají klinické studie cílené terapie proti uvedenému receptoru (např. NCT01560260)(10,31). Zdá se, že v cílené terapii GIST bude v budoucnu vhodnější a účinnější vzájemná kombinace terapie TKI (monoterapie TKI není dostatečná), společně s jinými léčivy (např. kombinace inhibitoru PI3K a IM – zejména u bodové mutace D842V, která je rezistentní na IM)(43), popřípadě imunoterapie (ipilimumab). Možnosti ovlivnění GIST se během několika let značně rozšířili a lze očekávat, že cílená terapie bude při kombinaci léčiv mnohem účinnější než dosavadní terapeutické přístupy. Precizní molekulární diagnostika GIST bude mít i nadále svou roli při indikaci přiměřených kombinací biologických léků.
MOLEKULÁRNÍ DIAGNOSTIKA GIST - SOUČASNÝ STAV
Zdrojem materiálu pro molekulární analýzu mutačního stavu genů KIT a PDGFRA jsou vzorky primárních nádorů, recidiv nádorů nebo metastáz. Kvalita DNA má pro molekulárně genetické vyšetřování zcela zásadní a významnou roli. Nejvyšší kvalitu izolované DNA získáváme z nativního bioptického materiálu ve formě zmražených vzorků. U archivních vzorků dochází v procesu fixace ve formolu a zalévání do parafínu k degradaci DNA. Modifikace primární struktury DNA, fragmentace DNA a křížové vazby s proteiny znesnadňují izolaci DNA a následně negativně ovlivňuji průběh jednotlivých molekulárně genetických analýz. Přesto lze, s určitými omezeními molekulární analýzu provést i z formolem fixované tkáně. Kontrola kvality a integrity izolované DNA, společně s volbou vhodných reakčních podmínek je nevyhnutnou součástí mutační analýzy GIST.
Přítomnost primárních mutací vyšetřujeme v exonech 9, 11, 13, a 17 genu KIT a v exonech 12, 14, a 18 genu PDGFRA. Mutace ve všech uvedených exonech analyzujeme najednou. Každou detekovanou mutaci ověřujeme opětovnou nezávislou amplifikací s následným zjištěním pořadí nukleotidů pomocí kapilární elektroforézy na genetickém analyzátoru. V případě, že se v analyzovaných exonech mutace nevyskytují, můžeme mutační analýzu rozšířit o zjištění přítomnosti mutací v exonu 8 genu KIT. U pacientů s GIST bez mutací v genech KIT a PDGFRA pokračujeme v mutační analýze genu BRAF. Konkrétně vyšetřujeme exony 11 a 15 uvedeného genu. Nejprve vyšetřujeme exon 15 genu BRAF, kde se mutace vyskytují nejčastěji. Následně doplníme mutační analýzu o vyšetření exonu 11 genu BRAF. U KIT/PDGFRA nemutovaných GIST mohou být v ojedinělých případech mutace přítomny rovněž v genech KRAS (event. NRAS) nebo PI3K. Mutační stav těchto genů vyšetřujeme jen v případech GIST, u kterých nejsou zjištěny mutace genů KIT, PDGFRA a BRAF.
V menší míře se u sporadických GIST s nemutovanými geny KIT/PDGFRA, případně u GIST, které tvoří součást syndromu Carney-Stratakis setkáváme s defektem a ztrátou funkce SDH komplexu. Defekt SDH komplexu je způsoben přítomností mutací v genech SDHA, SDHB, SDHC, nebo SDHD. Na analýze mutačního stavu genů SDH komplexu spolupracujeme s Klinikou dětské hematologie a onkologie 2. LF UK a FN Motol.
U pacientů s progresí onemocnění na terapii IM vyšetřujeme přítomnost sekundárních mutací. Opětovně analyzujeme vzorky, u kterých byly zjištěny primární mutace. Neprovádíme ovšem detekci mutací ve všech exonech genů KIT a PDGFRA. Vyšetřujeme pouze exon s přítomnou primární mutací a exony 13 a 17 genu KIT, kde se sekundární mutace vyskytují nejčastěji. V případě, že byla primární mutace zjištěna v některém z exonu genu PDGFRA, analyzujeme pouze gen PDGFRA. Za podmínky, že mutace v exonech 13 a 17 nedetekujeme, rozšiřujeme mutační analýzu o vyšetření přítomnosti mutací v exonech 14 a 18 genu KIT. Analýzu sekundární rezistence nádorů bez přítomnosti sekundárních mutací v exonech 13, 14, 17, a 18 genu KIT doplňujeme o zjištění mutačního stavu genu BRAF. Molekulární analýzu sekundárních mutací s ohledem na jejich velkou heterogenitu „v“ i „mezi“ jednotlivými metastatickými ložisky vždy provádíme ze všech dostupných bioptických vzorků.
Správně a precizně provedená mutační analýza se neobejde bez výběru vhodného vzorku, určení procentuálního zastoupení nádorových buněk ve vyšetřovaném materiálu, případně posouzení nenádorové příměsi či nekrotických ložisek, které významně ovlivňují výsledek molekulárního vyšetření. Přesnost molekulární diagnostiky GIST tak velmi významně závisí na úzké spolupráci s patologem.
PODĚKOVÁNÍ
Podpořeno projektem (Ministerstva zdravotnictví) koncepčního rozvoje výzkumné organizace 00064203 (FN MOTOL) - IG VZ 6006 a OPPK CZ.2.16/3.1.00/24022.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
Mgr. Alena Kalfusová
Ústav patologie a molekulární medicíny 2. LF UK a
FN Motol
V Úvalu 84,
150 06 Praha 5 - Motol
tel.: 224 435 622, 224 435 650
email: alena.kalfusova@fnmotol.cz
Zdroje
1. Ordog T, Zörnig M, Hayashi Y. Targeting disease persistance in gastrointestinal stromal tumors. Stem Cells Trans Med 2015; 4: 702-707.
2. Corless CL. Gastrointestinal stromal tumors: what do we know now? Mod Pathol 2014; 27: S1-S16.
3. Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit gene in human gastrointestinal stromal tumors. Science 1998; 279: 577-580.
4. Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299: 708-710.
5. Drucker BJ, Tamura S, Buchdunker E, et al. Effects of a selective inhibitor of the abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature Medicine 1996; 2(5): 561-566.
6. Al Ali HK, Heinrich MC, Lange T, et al. High incidence of BCR-ABL kinase domain mutations and absence of mutations of the PDGFR and KIT activation loops in CML patiens with secondary resistance to imatinib. Hematol J 2004; 5(1): 55-60.
7. Gorre ME, Mohhamed M, Ellwood K, et al. Clinical resistence to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293: 876-880.
8. Antonescu CR, Besmer P, Guo T, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res 2005; 11: 4182-4190.
9. Augustiňáková A, Kodet R. Molekulární diagnostika gastrointestinálních stromálních tumorů ve vztahu k predikci terapeutické odpovědi na cílenou biologickou léčbu. Cesk Patol 2011; 47(4): 148-152.
10. Gounder MM, Maki RG. Molecular basis for primary and secondary tyrosine kinase inhibitor resistance in gastrointestinal stromal tumor. Cancer Chemother Pharmacol 2011; 67(Suppl 1): 25-43.
11. Heinrich MC. Molecular basis for treatment of gastrointestinal stromal tumours. EJC Supplements 2006; 10-16.
12. Nishida T, Kanda T, Nishitani A, et al. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci 2008; 99(4): 799-804.
13. Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res 2009; 15(24): 7510-7518.
14. Agaram NP, Besmer P, Wong GC, et al. Pathologic and molecular heterogenity in imatinib-stable or imatinib-responsive gastrointestinal stromal tumors. Clin Cancer Res 2007; 13: 170-181.
15. Wardelmann E, Thomas N, Merkelbach-Bruse S, et al. Acquired resistance to imatinib in gastrointestinal stromal tumors caused by multiple KIT mutations. Lancet Oncol 2005; 6: 249-251.
16. Liegl B, Kepten I, Le C, et al. Heterogenity of kinase inhibitor resistance mechanisms in GIST. J Pathol 2008; 216(1): 64-74.
17. Wardelmann E, Merkelbach-Bruse S, Pauls K, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res 2006; 12(6): 1743-1749.
18. Maleddu A, Pantaleo MA, Nannini M, et al. Mechanisms of secondary resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors (Review). Oncology Reports 2009; 21: 1359-1366.
19. Antonescu CR, Romeo S, Zhang L, et al. Dedifferentiation in gastrointestinal stromal tumor to an anaplastic KIT negative phenotype – a diagnostic pitfall. Morphologic and molecular characterization of 8 cases occuring either de-novo or after imatinib therapy. J Am Surg Pathol 2013; 37(3): 385-392.
20. Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008; 47(10): 853-859.
21. Zheng S, Huang K, Jia J, et al. Rhabdomyosarcomatous differentiation in gastrointestinal stromal tumors after imatinib resistance: a potential diagnostic pitfall. Exp Biol Med 2013; 238: 120-124.
22. Nannini M, Astolfi A, Urbini M, et al. Integrated genomic study of quadruple-wt GIST (KIT/PDGFRA/SDH/RAS pathway wild type GIST). BMC Cancer 2014; 14: 685.
23. Daum O, Šedivcová M. Gastrointestinální stromální tumor (GIST): Pokroky do roku 2013. Cesk Pathol 2014; 50(2): 76-80.
24. Hostein I, Faur N, Primois Ch, et al. BRAF mutation status in gastrointestinal stromal tumors. Am J Clin Pathol 2010; 133: 141-148.
25. Rossi S, Sbaraglia M, Campo Dell´Orto M, et al. Concomitant KIT/BRAF and PDGFRA/BRAF mutations are rare events in gastrointestinal stromal tumors. Oncotarget 2016; 7(21): 30109-30118.
26. Agaimy A, Terracciano LM, Dirnhofer S, et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumor. J Clin Pathol 2009; 62: 613-616.
27. Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res 2012; 18(6): 1769-1776.
28. Daniels M, Lurkin I, Pauli R, et al. Spectrum of KIT/PDGFRA/BRAF mutations and phosphatydilinositol-3-kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett 2011; 312(1): 43-54.
29. Belinsky MG, Skorobogatko YV, Rink L, et al. High density DNA array analysis reveals distinct genomic profiles in a subset of gastrointestinal stromal tumors. Genes Chromosomes Cancer 2009; 48(10): 886-896.
30. Lasota J, Xi L, Coates T, et al. No KRAS mutations found in gastrointestinal stromal tumors (GISTs): molecular genetic study of 514 cases. Mod Pathol 2013; 26: 1488-1491.
31. Beadling C, Patterson J, Justusson E, et al. Gene expression of the IGF pathway family distinguishes subsets of gastrointestinal stromal tumors wild type or KIT and PDGFRA. Cancer Med 2013; 2(1): 21-31.
32. Miettinen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) – a review. Int J Biochem Cell Biol 2014; 53: 514-519.
33. Wang YM, Gu Ml, Ji F. Succinate dehydrogenase-deficient gastrointestinal stromal tumors. World J Gastroenterol 2015; 21(8): 2303-2314.
34. Tornillo L. Gastrointestinal stromal tumor – envolving concept. Front Medicine 2014; 1:43.
35. Pantaleo MA, Astolfi A, Urbini M, et al. Analysis of all subunits, SDHA, SDHB, SDHC, and SDHD of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet 2014; 22(1): 32-39.
36. Celestino L, Lima J, Faustino A, et al. Molecular alterations and expression of succinate dehydrogenase complex in wild-type KIT/PDGFRA/BRAF gastrointestinal stromal tumors. Eur J Hum Genet 2013; 21(5): 503-510.
37. Lasota J, Wang Z, Kim SY, et al. Expression of the receptor type 1 insuline-like growth factor (IGF1R) in gastrointestinal stromal tumor. An immunohistochemical study of 1078 cases with diagnostic and therapeutic implications. Am J Surg Pathol 2013; 37(1): 114-119.
38. Janeway KA, Zhu MJ, Barretina J, et al. Strong expression of IGF1R in pediatric gastrointestinal stromal tumors without IGF1R genomic amplification. Int J Cancer 2010; 127(11): 2718-2722.
39. Belinski MG, Rink L, Cai KQ, et al. Somatic loss of function mutations in neurofibromin 1 and MYC associated factor X genes identified by exome-wide sequencing in a wild-type GIST case. BMC Cancer 2015; 15: 887.
40. Zhao X, Yue Ch. Gastrointestinal stromal tumor. J Gastrointest Oncol 2012; 3(3): 189-208.
41. Bajciova V. Pediatrický GIST. Onkologie 2016; 10(5): 218-223.
42. Garner AP, Gozgit JM, Anjum R, et al. Ponatinib inhibits polyclonal drug-resistent KIT oncoproteins and shows therapeutic potential in heavily pretreated gastrointestinal stromal tumor (GIST) patients. Clin Cancer Res 2014; 20(22): 5745-5755.
43. Schroeder B, Li Z, Cranmer LD, et al. Targeting gastrointestinal stromal tumors: the role of regorafenib. Onco Targets Ther 2016; 9: 3009-3016.
44. George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol 2012; 30(19): 2401-2407.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2017 Číslo 4
Nejčtenější v tomto čísle
- Co nového v Ewing-like family aneb malobuněčné/kulatobuněčné sarkomy měkkých tkání a kostí s rearanží genů CIC a BCOR. Přehled problematiky a naše prvotní zkušenosti
- Molekulární patologie plicních karcinomů pro rutinní praxi – update 2017
-
Jaká je Vaše diagnóza? (1)
Odpověď - Molekulární mechanizmy primární a sekundární rezistence, molekulárně-genetické znaky a vlastnosti KIT/PDGFRA nemutovaných GIST